



RT-qPCR از فناوری PCR معمولی توسعه یافته است.این مواد شیمیایی فلورسنت (رنگ های فلورسنت یا پروب های فلورسنت) را به سیستم واکنش سنتی PCR اضافه می کند و فرآیند بازپخت و گسترش PCR را در زمان واقعی با توجه به مکانیسم های مختلف شب تاب آنها تشخیص می دهد.تغییرات سیگنال فلورسنت در محیط برای محاسبه مقدار تغییر محصول در هر سیکل PCR استفاده می شود.در حال حاضر رایج ترین روش ها روش رنگ فلورسنت و روش پروب می باشد.
روش رنگرزی فلورسنت:
برخی از رنگ های فلورسنت مانند SYBR Green Ⅰ، PicoGreen، BEBO و غیره به خودی خود نور ساطع نمی کنند، اما پس از اتصال به شیار کوچک dsDNA، فلورسانس ساطع می کنند.بنابراین، در ابتدای واکنش PCR، دستگاه نمی تواند سیگنال فلورسنت را تشخیص دهد.هنگامی که واکنش به مرحله بازپخت - گسترش (روش دو مرحلهای) یا مرحله گسترش (روش سه مرحلهای) ادامه میدهد، رشتههای دوتایی در این زمان باز میشوند و DNA پلیمراز جدید در طول سنتز رشته، مولکولهای فلورسنت در شیار کوچک dsDNA ترکیب میشوند و فلورسانس منتشر میکنند.با افزایش تعداد چرخه های PCR، رنگ های بیشتری با dsDNA ترکیب می شوند و سیگنال فلورسنت نیز به طور مداوم افزایش می یابد.به عنوان مثال SYBR Green Ⅰ را در نظر بگیرید.
روش پروب:
پروب Taqman متداول ترین پروب هیدرولیز مورد استفاده است.یک گروه فلورسنت در انتهای 5 دقیقه پروب وجود دارد که معمولاً FAM است.خود پروب یک توالی مکمل ژن هدف است.یک گروه خاموش کننده فلورسنت در انتهای 3' فلوروفور وجود دارد.با توجه به اصل انتقال انرژی تشدید فلورسانس (انتقال انرژی تشدید فورستر، FRET)، هنگامی که گروه فلورسنت گزارشگر (مولکول فلورسنت دهنده) و گروه فلورسنت خاموش کننده (مولکول فلورسنت گیرنده) زمانی که طیف تحریک با هم همپوشانی داشته باشد و فاصله می تواند بسیار نزدیک باشد (7-10). فلورسانس مولکول پذیرنده، در حالی که اتوفلورسانس ضعیف شده است.بنابراین، در ابتدای واکنش PCR، زمانی که پروب در سیستم آزاد و دست نخورده است، گروه فلورسنت گزارشگر فلورسانس منتشر نخواهد کرد.هنگام بازپخت، پرایمر و پروب به قالب متصل می شوند.در طول مرحله گسترش، پلیمراز به طور مداوم زنجیره های جدید را سنتز می کند.DNA پلیمراز دارای فعالیت اگزونوکلئاز 5'-3 است.هنگام رسیدن به پروب، DNA پلیمراز پروب را از قالب هیدرولیز می کند، گروه فلورسنت گزارشگر را از گروه فلورسنت خاموش کننده جدا می کند و سیگنال فلورسنت را آزاد می کند.از آنجایی که بین پروب و قالب رابطه یک به یک وجود دارد، روش پروب از نظر دقت و حساسیت آزمایش بر روش رنگرزی برتری دارد.
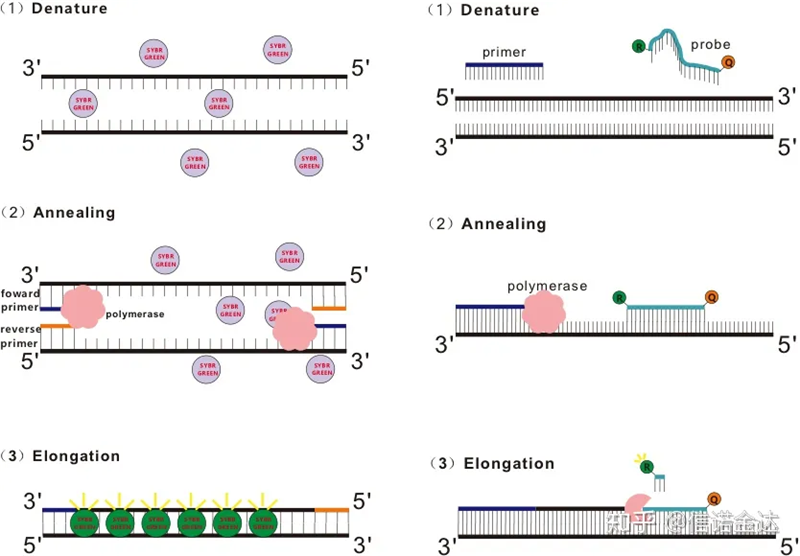
شکل 1 اصل qRT-PCR
طراحی پرایمر
اصول:
پرایمرها باید در ناحیه حفاظت شده سری نوکلئیک اسید طراحی شده و دارای ویژگی باشند.
بهتر است از توالی cDNA استفاده شود و توالی mRNA نیز قابل قبول است.اگر نه، طراحی ناحیه cds توالی DNA را پیدا کنید.
طول محصول کمی فلورسنت 80-150 جفت باز است، طولانی ترین آن 300 جفت باز است، طول پرایمر به طور کلی بین 17-25 پایه است و تفاوت بین پرایمرهای بالادست و پایین دست نباید خیلی زیاد باشد.
محتوای G+C بین 40 تا 60 درصد است و 45 تا 55 درصد بهترین است.
مقدار TM بین 58-62 درجه است.
سعی کنید از دایمرهای پرایمر و خود دایمرها اجتناب کنید، (بیش از 4 جفت پایه مکمل متوالی به نظر نرسد) ساختار سنجاق سر، در صورت اجتناب ناپذیر، ΔG<4.5kJ/mol بسازید* اگر نمی توانید اطمینان حاصل کنید که gDNA در حین رونویسی معکوس پاک شده است، بهتر است پرایمرهای *3T، انتهای G، اصلاح شده، C را طراحی کنید تا از آن جلوگیری شود. /C، ساختار پیوسته A/G (2-3) آغازگرها و غیر
خاص همسانی توالی ناهمگن تقویت شده ترجیحاً کمتر از 70 درصد است یا دارای 8 همولوژی پایه مکمل است.
پایگاه داده:
جستجوی CottonFGD با کلمات کلیدی
طراحی پرایمر:
طراحی پرایمر IDT-qPCR
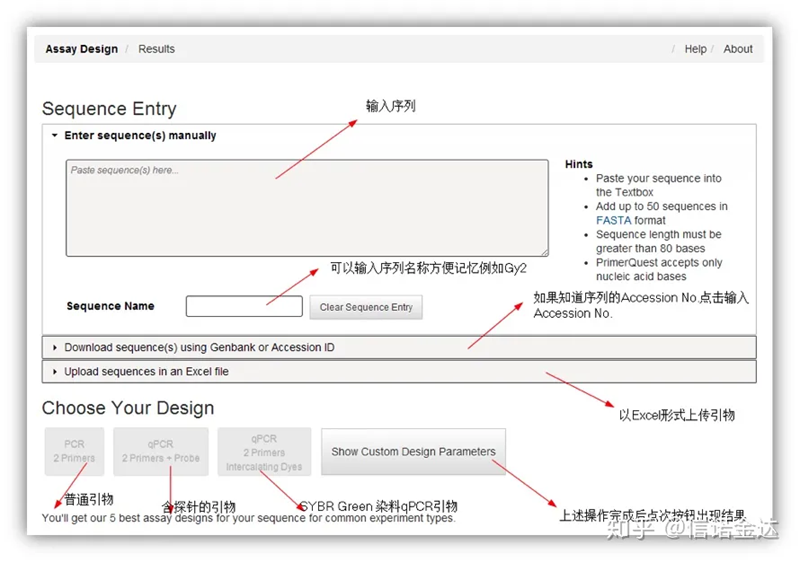
صفحه ابزار طراحی پرایمر آنلاین Fig2 IDT
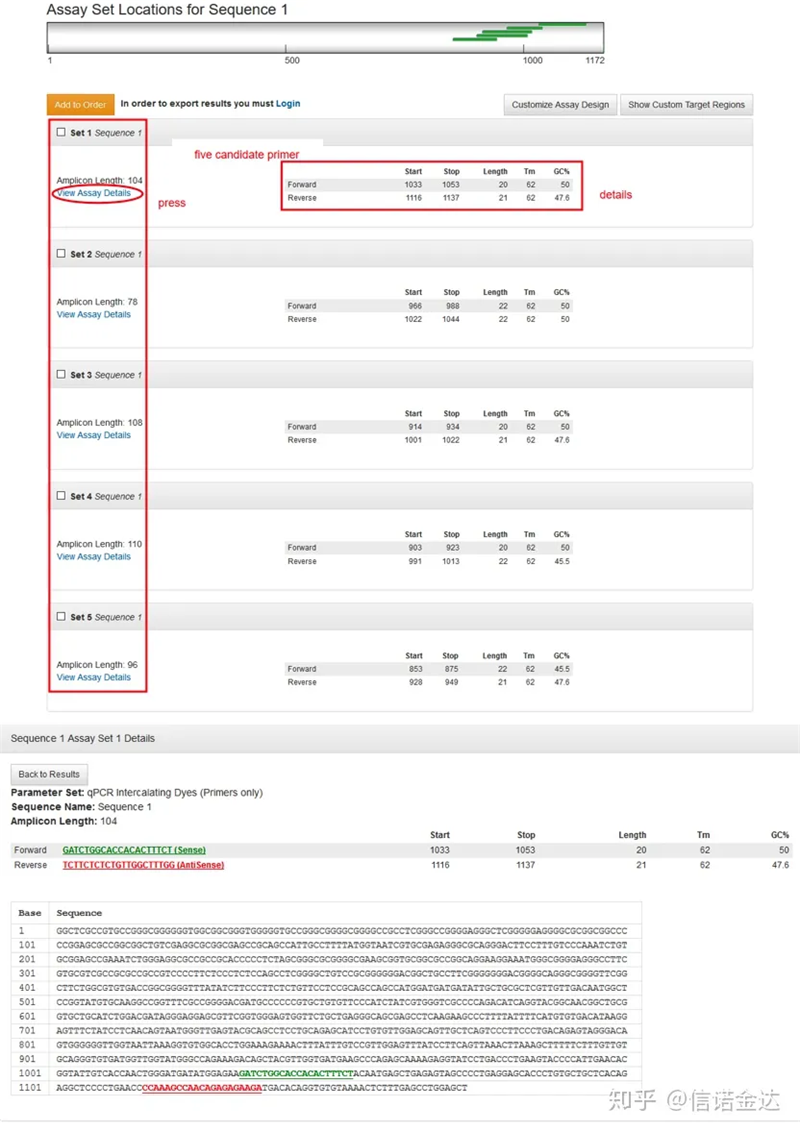
Fig3 نمایش صفحه نتیجه
طراحی پرایمرهای lncRNA:
lncRNA:همان مراحل mRNA
miRNA:اصل روش stem-loop: از آنجایی که همه miRNA ها توالی کوتاهی در حدود 23 nt هستند، تشخیص مستقیم PCR را نمی توان انجام داد، بنابراین از ابزار stem-loop sequence استفاده می شود.توالی حلقه ساقه یک DNA تک رشته ای با حدود 50 nt است که می تواند به خودی خود یک ساختار سنجاق مو را تشکیل دهد.3 'انتها را می توان به عنوان یک دنباله مکمل برای قطعه جزئی miRNA طراحی کرد، سپس miRNA هدف را می توان به دنباله حلقه ساقه در طول رونویسی معکوس متصل کرد و طول کل می تواند به 70bp برسد، که مطابق با طول محصول تقویت شده تعیین شده توسط qPCR است.طراحی پرایمر miRNA دنباله دار.
تشخیص ویژه تقویت:
پایگاه داده انفجار آنلاین: انفجار CottonFGD بر اساس شباهت توالی
Local blast: به استفاده از Blast+ برای انجام انفجار محلی مراجعه کنید، لینوکس و macos می توانند مستقیماً یک پایگاه داده محلی ایجاد کنند، سیستم win10 را نیز می توان پس از نصب اوبونتو bash انجام داد.ایجاد پایگاه داده انفجار محلی و انفجار محلی.اوبونتو bash را در win10 باز کنید.
توجه: پنبه مرتفع و پنبه جزیره دریایی محصولات تتراپلوئیدی هستند، بنابراین نتیجه انفجار اغلب دو یا چند کبریت خواهد بود.در گذشته، استفاده از سی دی های NAU به عنوان پایگاه داده برای انجام انفجار احتمالاً دو ژن همولوگ را با تنها چند تفاوت SNP پیدا می کند.معمولاً دو ژن همولوگ را نمیتوان با طراحی پرایمر از هم جدا کرد، بنابراین آنها را یکسان میدانیم.اگر ایندل واضح وجود داشته باشد، پرایمر معمولاً روی ایندل طراحی میشود، اما این ممکن است منجر به ساختار ثانویه پرایمر شود.
تشخیص ساختار ثانویه آغازگر:
مراحل:باز کردن اولیگو 7 ← توالی قالب ورودی ← بستن پنجره فرعی ← ذخیره ← مکان یابی پرایمر روی الگو، فشار دادن ctrl+D برای تنظیم طول پرایمر ← تجزیه و تحلیل ساختارهای ثانویه مختلف، مانند بدنه خود دیمریزاسیون، هترودایمر، سنجاق سر، عدم تطابق، و غیره. دو تصویر آخر در شکل 4 نتایج تست پرایمر هستند.نتیجه پرایمر جلو خوب است، ساختار دایمر و سنجاق سر واضحی وجود ندارد، پایه های مکمل پیوسته وجود ندارد و مقدار مطلق انرژی آزاد کمتر از 4.5 است، در حالی که پرایمر پشتی پیوسته را نشان می دهد. 6 پایه مکمل هستند و انرژی آزاد 8.8 است.علاوه بر این، یک دایمر جدی تر در انتهای 3 ظاهر می شود، و یک دایمر از 4 پایه متوالی ظاهر می شود.اگرچه انرژی آزاد زیاد نیست، دایمر 3' Chl می تواند به طور جدی بر ویژگی تقویت و راندمان تقویت تأثیر بگذارد.علاوه بر این، لازم است که سنجاق سر، هترودیمر و عدم تطابق را بررسی کنید.
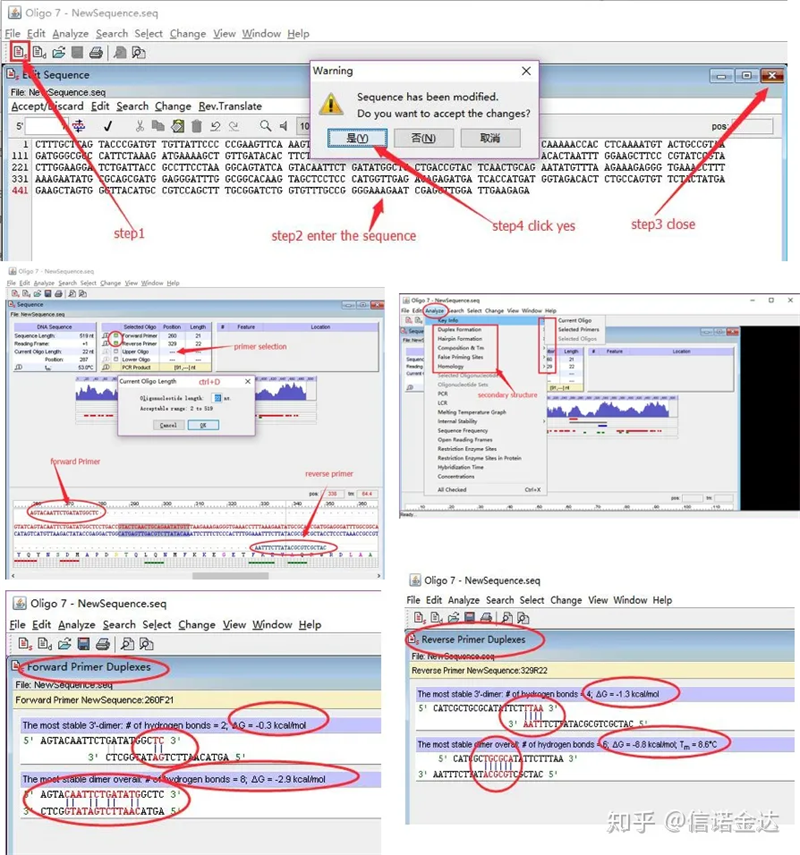
Fig3 نتایج تشخیص oligo7
تشخیص بازده تقویت:
راندمان تقویت واکنش PCR به طور جدی بر نتایج PCR تأثیر می گذارد.همچنین در qRT-PCR، راندمان تقویت به ویژه برای نتایج کمی مهم است.سایر مواد، ماشین ها و پروتکل ها را در بافر واکنش حذف کنید.کیفیت پرایمرها نیز تأثیر زیادی بر راندمان تقویت qRT-PCR دارد.برای اطمینان از صحت نتایج، هر دو کمیت فلورسانس نسبی و کمی فلورسانس مطلق نیاز به تشخیص بازده تقویت پرایمرها دارند.مشخص شده است که راندمان موثر تقویت qRT-PCR بین 85٪ و 115٪ است.دو روش وجود دارد:
1. روش منحنی استاندارد:
آ.cDNA را مخلوط کنید
برقیق شدن گرادیان
c.qPCR
دمعادله رگرسیون خطی برای محاسبه بازده تقویت
2. LinRegPCR
LinRegPCR برنامه ای برای تجزیه و تحلیل داده های RT-PCR زمان واقعی است که به داده های کمی PCR (qPCR) نیز بر اساس SYBR Green یا شیمی مشابه گفته می شود.این برنامه از دادههای اصلاحشده غیرپایه استفاده میکند، یک تصحیح پایه را روی هر نمونه به طور جداگانه انجام میدهد، یک پنجره خطی را تعیین میکند و سپس از تحلیل رگرسیون خطی برای جا دادن یک خط مستقیم در مجموعه دادههای PCR استفاده میکند.از شیب این خط راندمان PCR هر نمونه جداگانه محاسبه می شود.میانگین راندمان PCR در هر آمپلیکون و مقدار Ct در هر نمونه برای محاسبه غلظت اولیه در هر نمونه استفاده میشود که در واحدهای فلورسانس دلخواه بیان میشود.ورودی و خروجی داده ها از طریق صفحه گسترده اکسل می باشد.فقط نمونه
مخلوط کردن مورد نیاز است، بدون گرادیان
مراحل مورد نیاز است:(به عنوان مثال Bole CFX96 را در نظر بگیرید، نه کاملاً ماشینی با ABI واضح)
آزمایش:این یک آزمایش qPCR استاندارد است.
خروجی داده qPCR:LinRegPCR می تواند دو شکل از فایل های خروجی را تشخیص دهد: RDML یا نتیجه تقویت کمی.در واقع، این مقدار زمان واقعی تشخیص عدد سیکل و سیگنال فلورسانس توسط دستگاه است و تقویت با تجزیه و تحلیل مقدار تغییر فلورسانس بازده قطعه خطی به دست میآید.
انتخاب داده ها: در تئوری، مقدار RDML باید قابل استفاده باشد.تخمین زده می شود که مشکل کامپیوتر من این است که نرم افزار نمی تواند RDML را تشخیص دهد، بنابراین من مقدار خروجی اکسل را به عنوان داده اصلی دارم.توصیه می شود ابتدا یک غربالگری تقریبی از داده ها انجام شود، مانند عدم اضافه کردن نمونه و غیره، نقاط را می توان در داده های خروجی حذف کرد (البته نمی توانید آنها را حذف کنید، LinRegPCR در مرحله بعد این نکات را نادیده می گیرد)
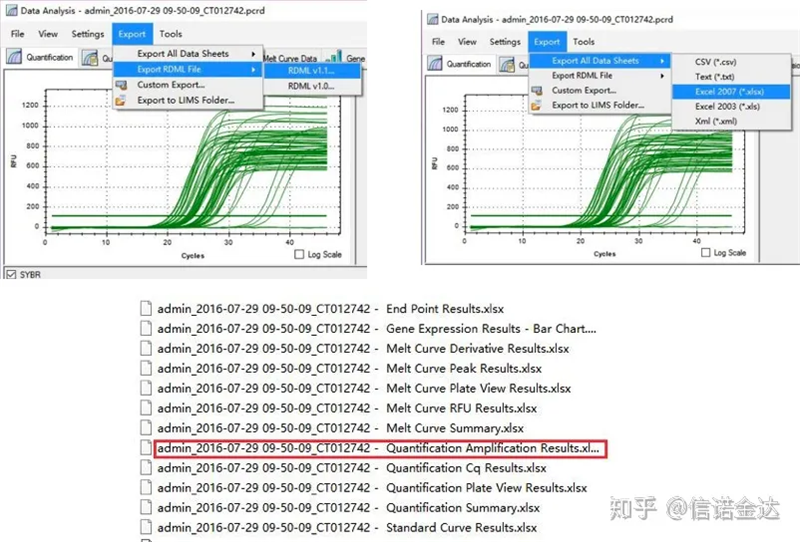
Fig5 صادرات داده qPCR
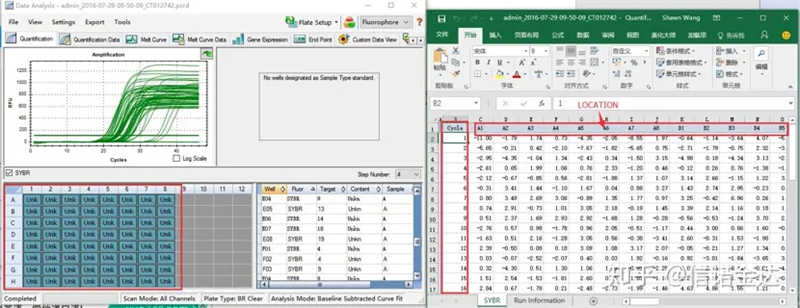
شکل 6 انتخاب نمونه های کاندید
ورود اطلاعات:باز کردن نتایج تقویت صلاحیت ها.
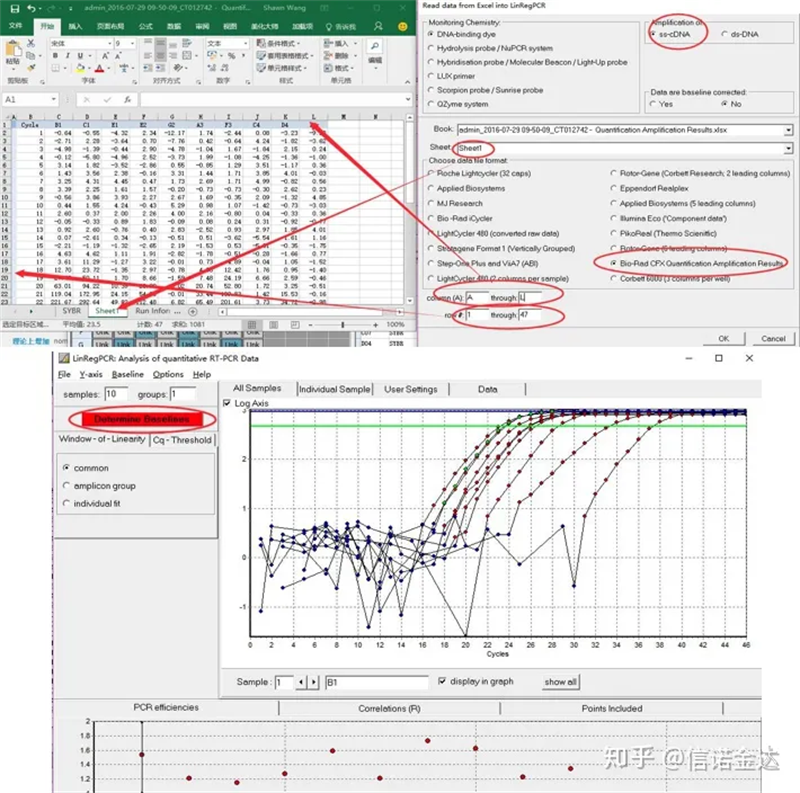
شکل 7 مراحل ورودی داده linRegPCR
نتیجه:در صورت عدم تکرار، نیازی به گروه بندی نیست.در صورت تکرار، می توان گروه بندی را در گروه بندی نمونه ویرایش کرد و نام ژن را در شناسه وارد کرد و سپس همان ژن به طور خودکار گروه بندی می شود.در نهایت روی فایل کلیک کرده و اکسل را صادر کرده و نتایج را مشاهده کنید.راندمان تقویت و نتایج R2 هر چاه نمایش داده می شود.ثانیاً، اگر به گروه ها تقسیم کنید، میانگین بازده تقویت اصلاح شده نمایش داده می شود.اطمینان حاصل کنید که راندمان تقویت هر پرایمر بین 85٪ و 115٪ است.اگر خیلی بزرگ یا خیلی کوچک باشد، به این معنی است که راندمان تقویت پرایمر ضعیف است.
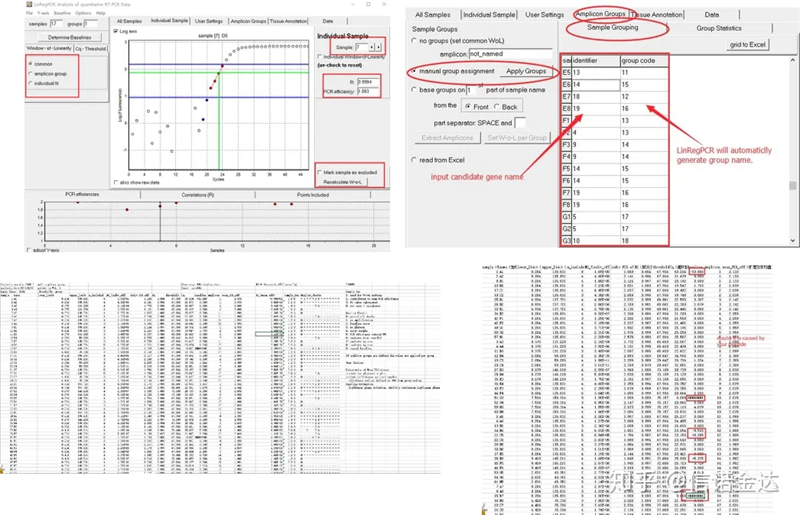
شکل 8 نتیجه و خروجی داده
فرآیند آزمایشی:
الزامات کیفیت RNA:
خلوص:1.72.0 نشان می دهد که ممکن است ایزوتیوسیانات باقی مانده وجود داشته باشد.اسید نوکلئیک تمیز A260/A230 باید حدود 2 باشد. اگر جذب قوی در nm 230 وجود داشته باشد، نشان دهنده وجود ترکیبات آلی مانند یون های فنات است.علاوه بر این، می توان آن را با الکتروفورز ژل آگارز 1.5٪ تشخیص داد.نشانگر را نشان دهید، زیرا ssRNA دناتوراسیون ندارد و لگاریتم وزن مولکولی رابطه خطی ندارد و وزن مولکولی را نمی توان به درستی بیان کرد.تمرکز: از نظر تئورینهکمتر از 100ng/ul، اگر غلظت خیلی کم باشد، خلوص به طور کلی کم است نه بلند
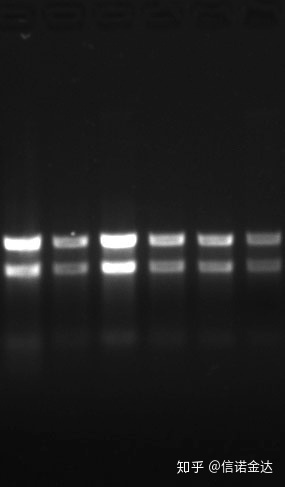
ژل RNA Fig9
علاوه بر این، در صورتی که نمونه گرانبها و غلظت RNA بالا باشد، توصیه می شود پس از استخراج، آن را به مقدار کافی تقسیم کنید و برای رونویسی معکوس، RNA را تا غلظت نهایی 100-300ng/ul رقیق کنید.که درفرآیند رونویسی معکوسهنگامی که mRNA رونویسی می شود، پرایمرهای الیگو (dt) که می توانند به طور خاص به دنباله های polyA متصل شوند، برای رونویسی معکوس استفاده می شوند، در حالی که lncRNA و circRNA از پرایمرهای تصادفی هگزامر (Random 6 mer) برای رونویسی معکوس RNA کل برای miRNA، miRNA-primers اختصاصی گردن برای rescription-loopverse استفاده می شود.در حال حاضر بسیاری از شرکت ها کیت های باطله ویژه را راه اندازی کرده اند.برای روش stem-loop، روش tailing راحتتر، بازده بالا و صرفهجویی در معرف است، اما تأثیر تمایز miRNAهای یک خانواده نباید به خوبی روش Stem-Loop باشد.هر کیت رونویسی معکوس دارای الزاماتی برای غلظت پرایمرهای اختصاصی ژن (حلقه های ساقه) است.مرجع داخلی مورد استفاده برای miRNA U6 است.در فرآیند وارونگی ساقه-حلقه، یک لوله U6 باید به طور جداگانه معکوس شود و پرایمرهای جلو و عقب U6 باید مستقیماً اضافه شود.هم circRNA و هم lncRNA می توانند از HKG به عنوان مرجع داخلی استفاده کنند.که درتشخیص cDNA،
اگر مشکلی با RNA وجود ندارد، cDNA نیز باید خوب باشد.با این حال، اگر کمال آزمایش دنبال شود، بهتر است از یک ژن مرجع داخلی (ژن مرجع، RG) استفاده شود که بتواند gDNA را از cds متمایز کند.به طور کلی، RG یک ژن خانه داری است., HKG) همانطور که در شکل 10 نشان داده شده است.در آن زمان، پروتئین ذخیرهسازی سویا درست میکردم و از اکتین 7 حاوی اینترونها به عنوان مرجع داخلی استفاده میکردم.اندازه قطعه تکثیر شده این پرایمر در gDNA 452 جفت باز و اگر cDNA به عنوان الگو استفاده می شد، 142 جفت باز بود.سپس نتایج آزمایش نشان داد که بخشی از cDNA در واقع توسط gDNA آلوده شده است و همچنین ثابت کرد که در نتیجه رونویسی معکوس مشکلی وجود ندارد و می توان از آن به عنوان الگوی PCR استفاده کرد.اجرای مستقیم الکتروفورز ژل آگارز با cDNA بی فایده است و یک نوار منتشر است که قانع کننده نیست.
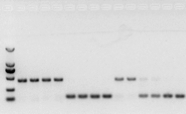
شکل 10 تشخیص cDNA
تعیین شرایط qPCRبه طور کلی با توجه به پروتکل کیت مشکلی وجود ندارد، عمدتاً در مرحله مقدار tm.اگر برخی از پرایمرها در طول طراحی پرایمر به خوبی طراحی نشده اند و در نتیجه اختلاف زیادی بین مقدار tm و 60 درجه سانتیگراد نظری ایجاد می شود، توصیه می شود که cDNA پس از مخلوط شدن نمونه ها، گرادیان PCR را با پرایمرها اجرا کنید و سعی کنید از تنظیم دما بدون باند به عنوان مقدار TM خودداری کنید.
تحلیل داده ها
روش مرسوم پردازش کمی فلورسانس نسبی PCR اساساً مطابق 2 است-ΔΔCT.الگوی پردازش داده ها
محصولات مرتبط:
Real Time PCR آسانTM -SYBR GREEN I
RT Easy I (Master Premix برای سنتز cDNA رشته اول)
RT Easy II (Master Premix برای سنتز cDNA رشته اول برای qPCR)
زمان ارسال: مارس-14-2023



