



1. درک اولیه
در این مرحله، ما باید مفاهیم و اصطلاحات را درک کنیم تا از انجام اشتباهات در مقابل سالمندان خود جلوگیری کنیم، مانند:
س: تفاوت بین RT-PCR، qPCR، Real-time PCR و Real-time RT-PCR چیست؟
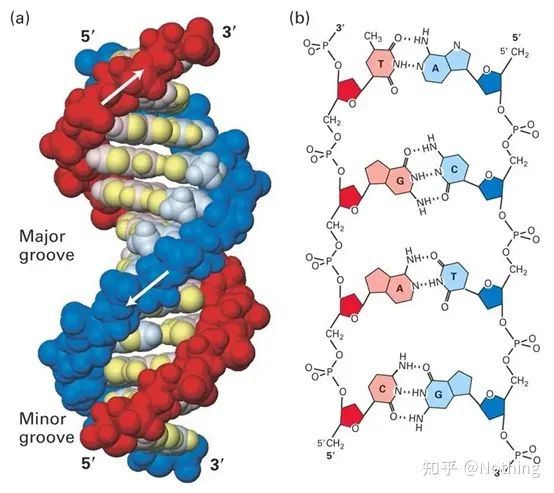
پاسخ: RT-PCR PCR رونویسی معکوس است(PCR رونویسی معکوس، RT-PCR)، که یک نوع پرکاربرد واکنش زنجیره ای پلیمراز (PCR) است.در RT-PCR، یک رشته RNA به DNA مکمل رونویسی معکوس می شود، که سپس به عنوان یک الگو برای تقویت DNA توسط PCR استفاده می شود.
Real-time-PCR و qPCR(Quantitative Rea-ltime-PCR) یک چیز هستند، هر دو PCR کمی زمان واقعی هستند، به این معنی که هر چرخه PCR دارای رکورد داده های بلادرنگ است، بنابراین می توان تعداد الگوهای شروع را تجزیه و تحلیل دقیق تنظیم کرد.
اگرچه به نظر می رسد هر دو Real-time PCR (Real-time fluorescent Quantitative PCR) و Reverse transcription PCR (PCR رونویسی معکوس) به اختصار RT-PCR هستند، کنوانسیون بین المللی این است: RT-PCR به طور خاص به رونویسی معکوس اشاره دارد.PCR، Real-time PCR به طور کلی به اختصار qPCR (کمی Real-time PCR) نامیده می شود..
و RT-PCR بلادرنگ (RT-qPCR)، این PCR رونویسی معکوس است که با فناوری کمی فلورسنت ترکیب شده است.: ابتدا cDNA (RT) را از رونویسی معکوس RNA بدست آورید و سپس از Real-time PCR برای تجزیه و تحلیل کمی (qPCR) استفاده کنید.اکثر آزمایشگاهها RT-qPCR انجام میدهند، یعنی تحقیق در مورد تنظیم پایین بیان RNA، بنابراین qPCR که همه در آزمایشگاه درباره آن صحبت میکنند در واقع به RT-qPCR اشاره دارد، اما فراموش نکنید که هنوز آزمایشهای DNA زیادی در کاربردهای بالینی وجود دارد.تجزیه و تحلیل کمی، مانند تشخیص ویروس هپاتیت B HBV.
سوال: پس از مطالعه زیاد PCR کمی فلورسنت، چرا قطعه تقویت شده باید در محدوده 80-300 جفت باز کنترل شود؟
پاسخ: طول هر توالی ژنی متفاوت است، برخی چندین کیلوبایت، برخی صدها جفت باز هستند، اما در طراحی پرایمرها فقط باید طول محصول 80-300 جفت باز باشد، خیلی کوتاه یا خیلی طولانی برای تشخیص کمی فلورسنت PCR مناسب نیستند.قطعه محصول خیلی کوتاه است که نمی توان آن را از پرایمر-دایمر متمایز کرد.طول پرایمر-دایمر حدود 30-40 جفت باز است و تشخیص اینکه پرایمر-دایمر است یا محصول اگر کمتر از 80 جفت باز باشد مشکل است.اگر قطعه محصول بیش از حد طولانی باشد، بیش از 300 جفت باز باشد، به راحتی منجر به راندمان تقویت پایین می شود و نمی تواند به طور موثر مقدار ژن را تشخیص دهد.
به عنوان مثال، وقتی شمارش می کنید که چند نفر در یک کلاس درس هستند، فقط باید تعداد دهان ها را بشمارید.هنگامی که ژنها را تشخیص میدهید، همین امر صادق است، فقط باید دنباله خاصی از یک ژن را شناسایی کنید تا کل توالی را نشان دهد.اگر می خواهید آدم ها را بشمارید باید هم دهان و هم بینی و گوش و عینک را بشمارید و اشتباه کردن آسان است.
برای گسترش، در تحقیقات بیولوژیکی، موارد تحقیقاتی زیادی از نقطهای به ناحیه دیگر وجود دارد، زیرا توالی ژنی هر گونه بسیار طولانی است، اندازهگیری همه قطعات غیرضروری و غیرممکن است، مانند توالییابی 16S باکتریایی، که عبارت است از انجام توالی محافظهکارانه باکتریها برای استنتاج تعداد جمعیت خاصی از باکتریها.
س: طول بهینه برای طراحی پرایمر qPCR چقدر است؟
پاسخ: به طور کلی طول پرایمر حدود 20-24 جفت باز است که بهتر است.البته در هنگام طراحی پرایمر باید به مقدار TM پرایمر توجه کنیم زیرا این امر مربوط به دمای بهینه آنیلینگ می باشد.پس از آزمایش های زیاد، ثابت شده است که 60 درجه سانتیگراد یک مقدار TM بهتر است.اگر دمای بازپخت خیلی پایین باشد، به راحتی منجر به تقویت غیر اختصاصی می شود.اگر دمای بازپخت بیش از حد بالا باشد، راندمان تقویت نسبتاً پایین خواهد بود، اوج منحنی تقویت دیرتر شروع می شود و مقدار CT به تأخیر می افتد.
س: روش رنگ با روش پروب چه تفاوتی دارد؟
جواب: روش رنگرزیبرخی از رنگ های فلورسنت مانند SYBR Green Ⅰ، PicoGreen، BEBO و غیره به خودی خود نور ساطع نمی کنند، اما پس از اتصال به شیار کوچک DNA دو رشته ای، فلورسانس منتشر می کنند.بنابراین، در ابتدای واکنش PCR، دستگاه نمی تواند سیگنال فلورسنت را تشخیص دهد.هنگامی که واکنش به مرحله بازپخت - گسترش می رسد، رشته دوتایی باز می شود و یک رشته جدید تحت عمل DNA پلیمراز سنتز می شود و مولکول فلورسنت به شیار کوچک dsDNA متصل می شود.با افزایش تعداد چرخه های PCR، رنگ های بیشتری با DNA دو رشته ای ترکیب می شوند و سیگنال فلورسنت نیز به طور مداوم افزایش می یابد.روش رنگ عمدتاً در تحقیقات علمی استفاده می شود.
PS: هنگام انجام آزمایش مراقب باشید، رنگ باید با DNA انسان ترکیب شود، مراقب باشید که آن را به یک فرد فلورسنت تبدیل کنید.
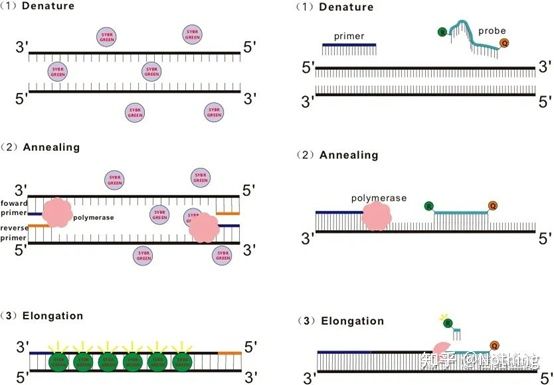
روش رنگرزی (چپ) روش پروب (راست)
PS: هنگام انجام آزمایش مراقب باشید، رنگ باید با DNA انسان ترکیب شود، مراقب باشید که آن را به یک فرد فلورسنت تبدیل کنید.
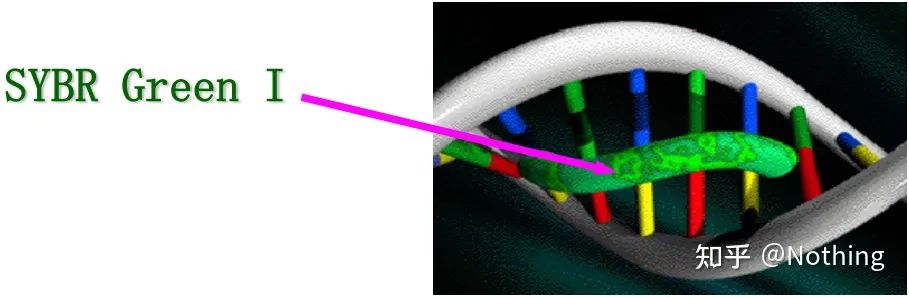
SYBR Green Ⅰ به شیار کوچک DNA متصل می شود
روش پروبپروب Taqman متداول ترین پروب هیدرولیز مورد استفاده است.یک گروه فلورسنت در انتهای 5 دقیقه پروب وجود دارد که معمولاً FAM است و خود پروب یک توالی مکمل ژن هدف است.یک گروه خاموش کننده فلورسنت در انتهای 3 دقیقه وجود دارد.با توجه به اصل انتقال انرژی تشدید فلورسانس (انتقال انرژی تشدید فورستر، FRET)، هنگامی که گروه فلورسنت گزارشگر (مولکول فلورسنت دهنده) و گروه فلورسنت خاموش کننده (مولکول فلورسنت گیرنده) برانگیخته می شوند، هنگامی که طیف ها همپوشانی دارند و فاصله می تواند بسیار نزدیک شود فلورسانس مولکول پذیرنده، در حالی که اتوفلورسانس ضعیف شده است.بنابراین، در ابتدای واکنش PCR، زمانی که پروب در سیستم آزاد و دست نخورده است، گروه فلورسنت گزارشگر فلورسانس منتشر نخواهد کرد.هنگام بازپخت، پرایمر و پروب به قالب متصل می شوند.در طول مرحله گسترش، پلیمراز به طور مداوم زنجیره های جدید را سنتز می کند.DNA پلیمراز دارای فعالیت اگزونوکلئاز 5'-3 است.هنگام رسیدن به پروب، DNA پلیمراز پروب را از قالب هیدرولیز می کند، گروه فلورسنت گزارشگر را از گروه فلورسنت خاموش کننده جدا می کند و سیگنال فلورسنت را آزاد می کند.از آنجایی که بین پروب و قالب رابطه یک به یک وجود دارد، روش پروب از نظر دقت و حساسیت آزمایش بر روش رنگرزی برتری دارد.روش پروب عمدتاً در تشخیص استفاده می شود.
س: کمیت مطلق چیست؟کمیت نسبی چیست؟
پاسخ: کمیت مطلق به محاسبه شماره کپی اولیه نمونه مورد آزمایش توسط qPCR اشاره دارد، مانند تعداد ویروس HBV در 1 میلی لیتر خون.نتیجه ای که با کمی سازی نسبی به دست می آید، تغییر در مقدار ژن هدف در یک نمونه خاص نسبت به نمونه مرجع دیگر است و بیان ژن با تنظیم بالا یا پایین تنظیم می شود.
س: آیا میزان استخراج RNA، راندمان رونویسی معکوس و کارایی تقویت بر نتایج تجربی تأثیر می گذارد؟
س: آیا ذخیره سازی نمونه، معرف های استخراج، معرف های رونویسی معکوس، و مواد مصرفی انتقال دهنده نور بر نتایج تجربی تأثیر می گذارد؟
س: چه روشی می تواند داده های تجربی را تصحیح کند؟
با توجه به این مسائل در قسمت های پیشرفته و پیشرفته زیر به طور مفصل به شرح آنها می پردازیم.
2. دانش پیشرفته
با توجه به PCR کمی فلورسنت زمان واقعی، ما باید این واقعیت را بشناسیم که هزاران مقاله تحقیقاتی علمی هر ساله منتشر می شود که در میان آنها فناوری PCR کمی فلورسنت تعداد کمی نیست.
اگر استاندارد مشترکی برای اندازه گیری آزمایش PCR کمی فلورسنت وجود نداشته باشد، نتایج ممکن است بسیار متفاوت باشد.برای یک ژن از همان گونه، با روش پردازش یکسان، نتایج تشخیص نیز بسیار متفاوت خواهد بود و تکرار نتایج مشابه برای افرادی که دیر وارد می شوند دشوار خواهد بود.شما هیچ کس نمی داند کدام درست است و کدام نادرست.
آیا این بدان معناست که PCR کمی فلورسنت یک فناوری تقلب است یا یک فناوری غیرقابل اعتماد؟خیر، به این دلیل است که PCR کمی فلورسنت حساستر و دقیقتر است و یک عمل کمی اشتباه نتایج کاملاً متضادی را به همراه خواهد داشت.یک ضرر کوچک هزار مایل دورتر است.نویسنده مقاله ممکن است بارها توسط داوران شکنجه شود.در عین حال، انتخاب داوران مجله نیز از بین نتایج تجربی مختلف دشوار است.
در مجموع، به عدم اجماع در آزمایشهای PCR بلادرنگ اشاره میکند.برای این منظور، دانشمندان ارشد در صنعت شروع به تدوین استانداردها کردند.از مشارکتکنندگان میخواهد تا برخی جزئیات آزمایشی و پردازش دادههای لازم (از جمله دادههای لازم) را در مقاله برای برآورده کردن این استانداردها ارائه کنند.
داوران می توانند با خواندن این جزئیات در مورد کیفیت آزمایش قضاوت کنند.خوانندگان آینده نیز می توانند از این برای تکرار آزمایش یا بهبود آزمایش استفاده کنند.سپس نتایج تجربی به دست آمده از این طریق پر از اطلاعات، کیفیت بالا و قابل استفاده است.
MIBBI (حداقل اطلاعات برای تحقیقات بیولوژیکی و زیست پزشکی -http://www.mibbi.org) به وجود آمد.MIBBI پروژه ای است که استانداردهایی را برای آزمایش ها ارائه می کند.در طبیعت منتشر می شود.این پروژه با هدف آزمایشهای بیولوژیکی مختلف از جمله زیستشناسی سلولی، میکروآرایه، qPCR که اکنون میخواهیم در مورد آن صحبت کنیم، و غیره است، و برای هر نوع آزمایش در هنگام ارسال نسخههای خطی ارائه میشود.این اطلاعات باید همیشه ارائه شود.
در پروژه MIBBI دو مقاله مربوط به PCR کمی فلورسنت وجود دارد.:
· RDML (زبان نشانه گذاری داده های PCR در زمان واقعی) - یک زبان ساختاریافته و راهنمای گزارش برای داده های کمی PCR در زمان واقعی.
·MIQE (حداقل اطلاعات برای انتشار آزمایش های کمی PCR در زمان واقعی) - حداقل اطلاعات برای انتشار مقالات در مورد آزمایش های کمی PCR در زمان واقعی.
ابتدا، اجازه دهید در مورد RDML، مشخصات اصطلاحات صحبت کنیم.
اگر تعریف استانداردی برای همه چیز وجود نداشته باشد، ادامه بحث غیرممکن است، به همین دلیل است که توضیح اصطلاحات در امتحان بسیار مهم است.
اصطلاحات مورد استفاده در آزمایش PCR کمی فلورسنت شامل محتوای زیر است.QIAGEN بهترین خلاصه را برای ما ساخته است.موارد زیر همگی خشک هستندکالاها .
منحنی تقویت
منحنی تقویت به منحنی ساخته شده در طول فرآیند PCR، با شماره چرخه به عنوان آبسیسا و شدت فلورسانس بلادرنگ در طول واکنش به عنوان اردین اشاره دارد.
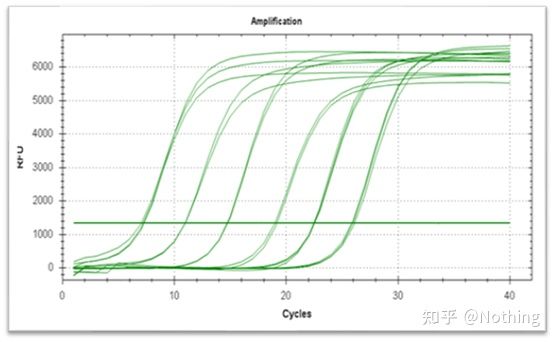
یک منحنی تقویت عالی باید ویژگی های زیر را داشته باشد: خط پایه صاف یا اندکی کاهش یافته است و روند صعودی آشکاری وجود ندارد.نقطه عطف منحنی واضح است و شیب فاز نمایی متناسب با بازده تقویت است.هرچه شیب بیشتر باشد، راندمان تقویت بیشتر است.منحنی تقویت کلی موازی سازی خوب است، که نشان می دهد بازده تقویت هر لوله مشابه است.فاز نمایی منحنی تقویت نمونههای با غلظت پایین واضح است.
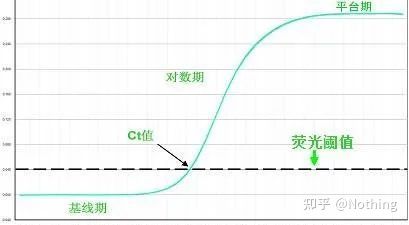
خط پایه (پایه)
خط پایه، سطح نویز سیکل اولیه استمعمولاً بین سیکل 3 و 15 اندازه گیری می شود، زیرا افزایش مقدار فلورسانس ناشی از محصول تقویت در این دوره قابل تشخیص نیست.تعداد چرخه های مورد استفاده برای محاسبه خط پایه می تواند متفاوت باشد و در صورت استفاده از مقادیر زیاد الگو یا اگر سطح بیان ژن هدف بالا باشد، ممکن است نیاز به کاهش داشته باشد.
تنظیم خط پایه مستلزم مشاهده داده های فلورسانس از منحنی تقویت خطی است.خط مبنا طوری تنظیم می شود که رشد منحنی تقویت با یک عدد سیکل بزرگتر از عدد بالای سیکل پایه شروع می شود.خطوط پایه باید به صورت جداگانه برای هر دنباله هدف تنظیم شوند.مقادیر متوسط فلورسانس شناسایی شده در چرخه های اولیه باید از مقادیر فلورسانس به دست آمده در محصولات تقویت شده کم شود.آخرین نسخههای مختلف نرمافزار Real-Time PCR امکان بهینهسازی خودکار تنظیمات پایه را برای نمونههای جداگانه فراهم میکند.
در طی چند سیکل اول واکنش تقویت PCR، سیگنال فلورسانس تغییر زیادی نمی کند.نزدیک شدن به یک خط مستقیم خط مبنا نامیده می شود، اما اگر به چند چرخه اول دقت کنیم، می بینیم که در داخل خط مبنا همان چیزی است که در تصویر زیر اتفاق می افتد.
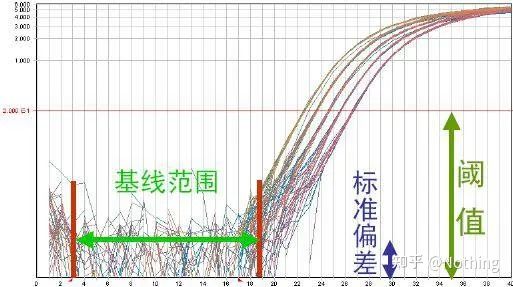
Background Background اشاره دارد به
مقدار فلورسانس غیر اختصاصی در واکنش.به عنوان مثال: خاموش کردن فلورسانس ناکارآمد.یا تعداد زیادی قالب DNA دو رشته ای به دلیل استفاده از SYBR Green.اجزای پس زمینه سیگنال به صورت ریاضی توسط الگوریتم نرم افزار Real-Time PCR حذف می شوند.
سیگنال گزارشگر
سیگنال گزارشگر به سیگنال فلورسنتی اطلاق می شود که توسط SYBR Green یا پروب های فلورسنت خاص توالی در طول Real-Time PCR تولید می شود.
سیگنال گزارشگر عادی (RN)
RN به شدت فلورسانس رنگ گزارشگر تقسیم بر شدت فلورسانس رنگ مرجع غیرفعال اندازه گیری شده در هر چرخه اشاره دارد.
رنگ مرجع غیرفعال
در برخی از Real-Time PCR ها،رنگ فلورسنت ROX به عنوان یک مرجع داخلی برای عادی سازی سیگنال فلورسنت استفاده می شود..تغییرات ناشی از لوله گذاری نادرست، موقعیت چاه و نوسانات فلورسانس را بر اساس چاه به چاه اصلاح می کند.

آستانه فلورسانس (آستانه)
بالاتر از مقدار پس زمینه و به طور قابل توجهی زیر مقدار فلات منحنی تقویت تنظیم شد.باید در ناحیه خطی منحنی تقویت قرار گیرد، که نشان دهنده محدوده لگاریتم خطی تشخیص PCR است.آستانه ها باید در نمای منحنی log-amplification تنظیم شوند تا فاز log-linear PCR به راحتی قابل شناسایی باشد.اگر چندین ژن هدف در Real-Time PCR وجود داشته باشد، آستانه باید برای هر هدف تنظیم شود.به طور کلی، سیگنال فلورسانس 15 سیکل اول واکنش PCR به عنوان سیگنال پسزمینه فلورسانس استفاده میشود و آستانه فلورسانس 10 برابر انحراف استاندارد سیگنال فلورسانس 3 تا 15 سیکل اول PCR است و فاز فلورسانس در مرحله PCR تنظیم میشود.به طور کلی، هر ابزار آستانه فلورسانس خود را قبل از استفاده تنظیم می کند.
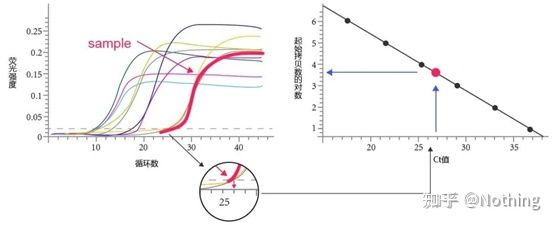
آستانه چرخه (CT) یا نقطه عبور (CP)
چرخه ای که در آن منحنی تقویت از آستانه عبور می کند (یعنی نقطه ای که در آن تشخیص فلورسانس به طور قابل توجهی افزایش می یابد).CT می تواند کسری باشد و مقدار الگوی شروع را می توان محاسبه کرد.مقدار CT نشان دهنده تعداد چرخه هایی است که هنگامی که سیگنال فلورسنت در هر لوله واکنش PCR به آستانه تنظیم شده می رسد تجربه می شود.یک رابطه خطی بین مقدار CT هر الگو و لگاریتم شماره کپی اولیه الگو وجود دارد.عدد کپی اولیه بالاتر، مقدار CT کوچکتر است و بالعکس.یک منحنی استاندارد را می توان با استفاده از یک استاندارد با شماره کپی اولیه شناخته شده ایجاد کرد که در آن ابسیسا نشان دهنده مقدار CT است و اردینات نشان دهنده لگاریتم شماره کپی اولیه است.بنابراین تا زمانی که مقدار CT نمونه مجهول به دست می آید، می توان شماره کپی اولیه نمونه را از روی منحنی استاندارد محاسبه کرد.
مقدار ΔCT
مقدار ΔCT توصیف می کندتفاوت بین ژن هدف و مقدار CT ژن مرجع درون زا مربوطهمانند ژن خانه داری، و برای عادی سازی مقدار الگوی استفاده شده استفاده می شود:
⇒ΔCT = CT (ژن هدف) - CT (ژن مرجع درون زا)
مقدار ΔΔCT
مقدار ΔΔCT تفاوت بین مقدار میانگین ΔΔCT نمونه مورد علاقه (به عنوان مثال، سلول های تحریک شده) و مقدار میانگین ΔΔCT نمونه مرجع (به عنوان مثال، سلول های تحریک نشده) را توصیف می کند.نمونه مرجع نیز نمونه کالیبراسیون نامیده می شود و سایر نمونه ها برای کمیت نسبی به این حالت نرمال می شوند:
⇒ΔΔCT = میانگین ΔCT (نمونه مورد علاقه) - متوسط ΔCT (نمونه مرجع)
ژن های مرجع درون زا (ژن های مرجع درون زا)
سطح بیان ژن های مرجع درون زا، مانند ژن های خانه داری (ژن های خانه داری)، بین نمونه ها تفاوتی ندارد.مقایسه مقادیر CT ژن مرجع با ژن هدف اجازه می دهد تا سطح بیان ژن هدف به مقدار RNA ورودی یا cDNA نرمال شود (به بخش مقادیر ΔCT در بالا مراجعه کنید).
ژن مرجع داخلی برایتخریب احتمالی RNA یا وجود مهارکنندههای آنزیم در نمونههای RNA، و همچنین تغییرات در محتوای RNA، راندمان رونویسی معکوس، بازیابی اسید نوکلئیک و جابجایی نمونه.برای انتخاب ژن(های مرجع) بهینه، الگوریتم را اصلاح کردیم تا امکان انتخاب مرجع بهینه وابسته به تنظیمات تجربی را فراهم کنیم.
کنترل داخلی
توالی کنترلی که در همان واکنش توالی هدف تقویت می شود و با یک پروب متفاوت کاوش می شود (یعنی انجام PCR دوبلکس).کنترل های داخلی اغلب برای رد تقویت های ناموفق استفاده می شود، مانند زمانی که دنباله هدف شناسایی نمی شود.
نمونه کالیبراسیون
یک نمونه مرجع (به عنوان مثال، RNA خالص از یک رده سلولی یا بافت) که در کمیت نسبی برای مقایسه تمام نمونه های دیگر برای تعیین سطح بیان نسبی یک ژن استفاده می شود.نمونه کالیبراسیون می تواند هر نمونه ای باشد، اما معمولاً یک کنترل است (به عنوان مثال، یک نمونه درمان نشده یا یک نمونه از زمان صفر آزمایش).
کنترل های مثبت
استفاده از واکنش های کنترلی بامقادیر شناخته شده الگو.کنترلهای مثبت اغلب برای بررسی اینکه مجموعه پرایمر یا مجموعه پرایمر-کاوشگر به درستی کار میکند و اینکه واکنش به درستی تنظیم شده است استفاده میشود.
بدون کنترل الگو (NTC)
یک واکنش کنترلی که شامل تمام اجزای ضروری واکنش تقویت به جز قالب است که معمولاً با آب جایگزین می شود.استفاده از NTC می تواند آلودگی ناشی از آلودگی معرف یا DNA خارجی را پیدا کند، بنابراین صحت و قابلیت اطمینان داده های تشخیص را تضمین می کند.تقویت کنترل NTC نشان دهنده آلودگی است.
بدون کنترل RT (NRT)
فرآیند استخراج RNA ممکن است حاوی DNA ژنومی باقیمانده باشد که بسیار مضر است و مقصری است که بر کیفیت دادهها و دشمن طبیعی qPCR تأثیر میگذارد، بنابراین هنگام طراحی آزمایشها، باید فقط به گونهای طراحی شود که تشخیص RNA را تقویت کند.دو راه وجود دارد، یکی طراحی پرایمرها در طول اینترون ها، دیگری حذف کامل DNA که کدام یک بهتر است، که بعداً مورد بحث قرار خواهد گرفت.کنترل NTR یک آینه جادویی برای تشخیص آلودگی DNA است.اگر تقویت باشد یعنی آلودگی وجود دارد.
استانداردها
استانداردها نمونه هایی از غلظت شناخته شده یا تعداد کپی هستند که برای ساخت منحنی استاندارد استفاده می شوند.به منظور اطمینان از پایداری استاندارد، قطعه ژن معمولاً در پلاسمید کلون شده و به عنوان استاندارد استفاده می شود.
منحنی استاندارد
معمولاً با محصول استاندارد با توجه به نسبت دو برابر شدن به حداقل 5 شیب غلظت رقیق می شود و 5 نقطه در مختصات مقدار CT و شماره کپی ترسیم می شود و نقاط برای تشکیل یک خط برای ایجاد یک منحنی استاندارد به هم متصل می شوند.برای هر منحنی استاندارد، اعتبار آن باید بررسی شود.مقدار شیب بین -3.3 و -3.8 قرار می گیرد و هر غلظت در سه تکرار انجام می شود.نقاطی که تفاوت قابل توجهی با سایر نقاط دارند باید کنار گذاشته شوند.مقدار CT نمونه مورد آزمایش به منحنی استاندارد آورده می شود و سطح بیان نمونه مورد آزمایش را می توان محاسبه کرد.
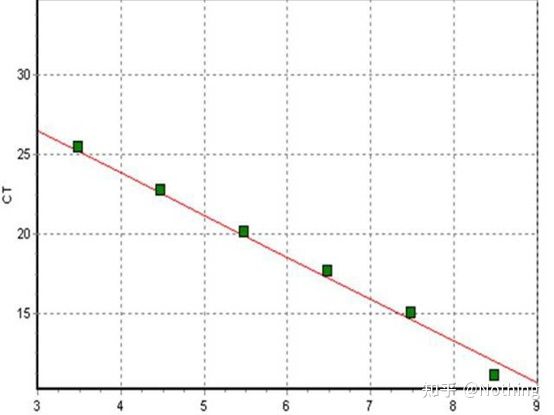
مقدار CT نمونه مورد آزمایش به منحنی استاندارد آورده می شود و می توان شماره کپی اولیه نمونه مورد آزمایش را محاسبه کرد.
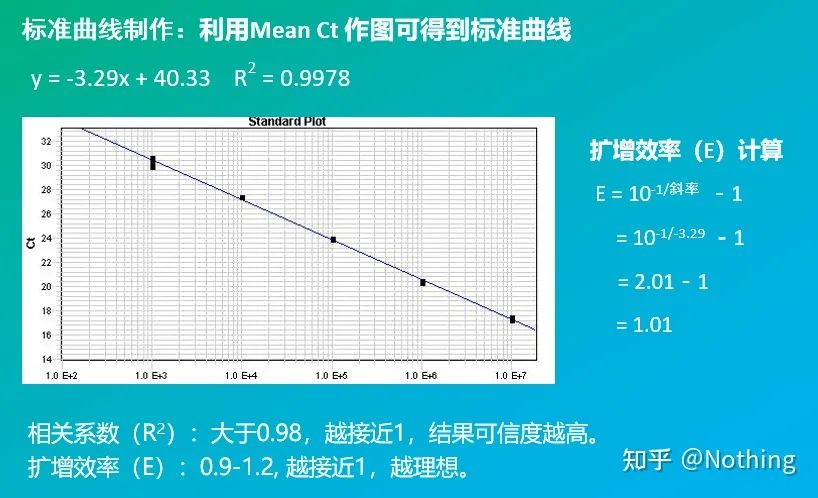
کارایی و شیب
شیب منحنی استاندارد نشان دهنده کارایی Real-time PCR است.
· شیب 3.322- نشان می دهد که راندمان تقویت PCR 1 یا 100٪ کارآمد است و مقدار محصول PCR در هر چرخه دو برابر می شود.
· شیب کمتر از 3.322- (مثلاً 3.8-) نشان دهنده کارایی PCR است.
· شیب بیشتر از -3.322 (به عنوان مثال، -3.0) نشان می دهد که به نظر می رسد راندمان PCR بیشتر از 100٪ است، که عجیب است، چگونه یک چرخه PCR می تواند بیش از دو برابر محصول تقویت شده تولید کند؟این وضعیت در فاز غیر خطی واکنش PCR رخ می دهد، یعنی مقدار زیادی تقویت غیر اختصاصی وجود دارد.
منحنی ذوب
پس از تکمیل تقویت qPCR، محصول PCR گرم می شود.با افزایش دما، محصول تقویت دو رشته ای به تدریج ذوب می شود و در نتیجه شدت فلورسانس کاهش می یابد.وقتی به دمای معینی (Tm) رسید، تعداد زیادی از محصولات ذوب می شوند.فلورسانس به شدت کاهش می یابد.محصولات مختلف PCR دارای مقادیر Tm متفاوت و دمای ذوب متفاوت هستند، به طوری که می توان ویژگی PCR را شناسایی کرد.
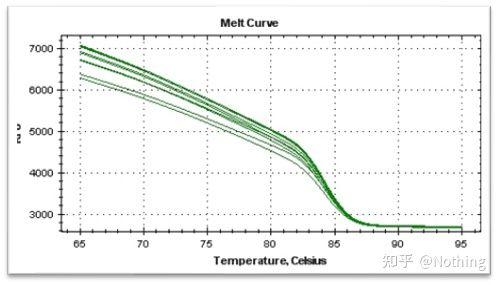
منحنی ذوب (منحنی مشتق)
منحنی ذوب برای تشکیل یک نقشه پیک به دست میآید که میتواند وضعیت قطعات محصول PCR را به طور مستقیم نشان دهد.از آنجایی که دمای ذوب مقدار Tm قطعه DNA است، برخی از پارامترهایی که بر مقدار Tm قطعه DNA تأثیر میگذارند میتوان قضاوت کرد، مانند اندازه قطعه، محتوای GC و غیره. به طور کلی، طبق اصول طراحی پرایمر ما،طول محصول تقویت شده در محدوده 80-300 جفت باز است، بنابراین دمای ذوب باید بین 80 تا 90 درجه سانتیگراد باشد.
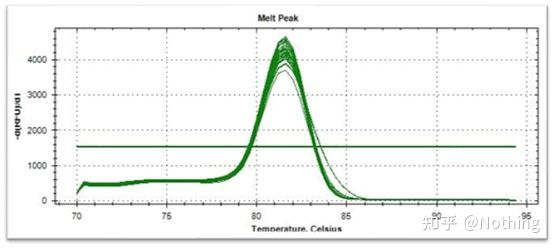
تفسیر منحنی ذوب: اگر تنها پیک اصلی بین 80-90 درجه سانتیگراد ظاهر شود، به این معنی است که PCR کمی فلورسنت عالی است.اگر پیک اصلی بین 80-90 درجه سانتیگراد ظاهر شود و پیک های متفرقه زیر 80 درجه سانتیگراد ظاهر شوند، اساساً دایمر آغازگر در نظر گرفته می شود.برای حل آن می توانید دمای بازپخت را افزایش دهید.اگر پیک اصلی بین 80-90 درجه سانتیگراد ظاهر شود، و پیک متفرقه دوباره با افزایش دما ظاهر شود، اساساً در نظر گرفته می شود که آلودگی DNA وجود دارد و DNA باید در مرحله اولیه آزمایش حذف شود.
البته هنوز موقعیت های غیرعادی وجود دارد که در ادامه یکی یکی به تفکیک آنها می پردازیم.
3. دانش پیشرفته
برای انجام qPCR، باید MIQE را بگویم،حداقل اطلاعاتبرای انتشارکمیReal-Time PCRآزمایش ها - حداقل اطلاعات برای انتشار مقالات در مورد PCR کمی زمان واقعیآزمایش .برای اینکه درک همه را ساده کنیم، محتوای کلیدی را ساده می کنیم.
شما می توانید متن اصلی MIQE را در اینترنت جستجو کنید و مهم ترین نکته این است که در آن قید شده استچک لیست داده هایی که باید هنگام انتشار مقاله ارائه شود .
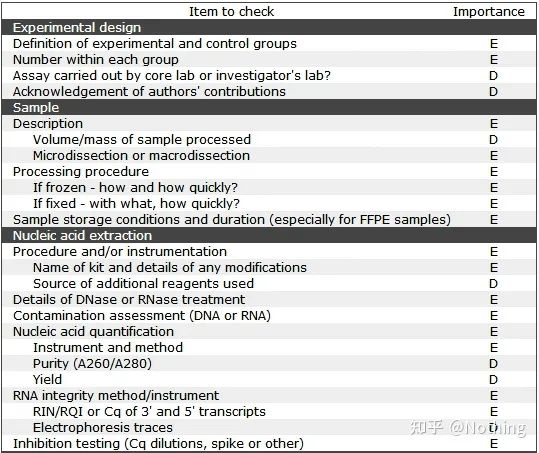
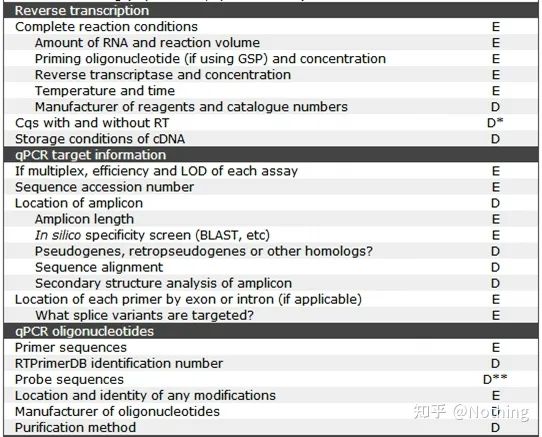
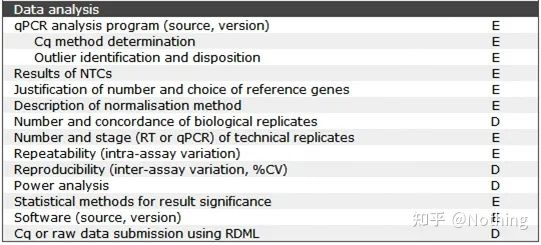
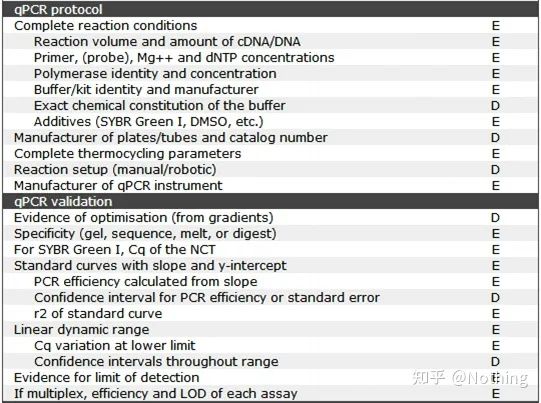
داوران می توانند با خواندن این جزئیات در مورد کیفیت آزمایش قضاوت کنند.خوانندگان آینده نیز می توانند از این برای تکرار یا بهبود آزمایش استفاده کنند.
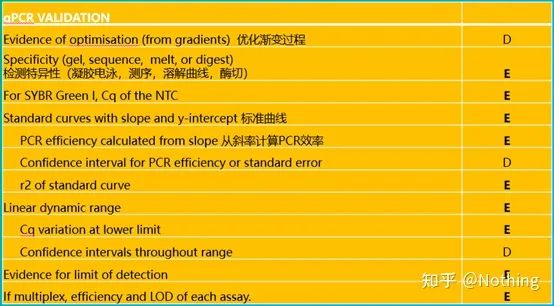
شایان ذکر است که در این لیست اهمیت هر لیست به ترتیب با E یا D مشخص شده است.چه مفهومی داره؟E: اطلاعات ضروری (باید ارسال شود)؛د: اطلاعات مطلوب (در حد امکان ارائه شود).
MIQE (1) - طراحی تجربی
بسیاری از شرورانی که پس از پایان تحصیلات تکمیلی دفاع خود را به پایان رسانده اند، نمی دانند چگونه آزمایشی را مستقل طراحی کنند، دفترچه های خود را باز کنند و آنچه معلم به آنها می گوید انجام دهند.در نتیجه، طراحی آزمایشی دقیق نبود و بخش تحریریه مجله گفت که میخواهند این عکس و آن عکس را بسازند، بنابراین با گیج این کار را انجام دادند.آشغال ها اینگونه درست می شوند!

نزدیکتر به خانه، اولین اصل آزمایش تعیین استسختگیری منطق تجربی.اساسی ترین چیز طراحی آزمایشی است و مهمترین چیز در مورد طرح آزمایشی نحوه تنظیم نمونه هدف، نمونه مرجع (شاهد) و تعداد تکرارها است تا داده های آزمایشی مرجع، قابل مقایسه و قانع کننده باشند.
نمونه هدفبه نمونه ای اطلاق می شود که ما را ملزم به شناسایی ژن هدف پس از یک درمان خاص می کند.نمونه مرجعنمونه بدون هیچ گونه درمان است که اغلب در زیست شناسی از آن به عنوان نوع وحشی یاد می شود.
تکرارهای آزمایشیبسیار مهم هستند.به طور کلی، تعداد تکرارهای متقاعد کننده باید بیش از سه باشد.باید تشخیص داد که تکثیر بیولوژیکی چیست و تکثیر فنی چیست.
تکرارهای بیولوژیکی: آزمایش تأیید یکسانی که با مواد مختلف (زمان، گیاهان، دسته ها، صفحات واکنش) انجام شده است.

تکثیر بیولوژیکی
بیایید درمان آفت کش فلفل را به عنوان مثال در نظر بگیریم.ما می خواهیم روی سه گیاه ABC سم پاشی کنیم، سپس سه گیاه ABC سه تکرار بیولوژیکی هستند و آنها همان آزمایش تأییدی هستند که با مواد مختلف انجام شده است.اما به عنوان یک آزمایش، قطعاً به یک شاهد نیاز است، بنابراین میتوانیم یکی از شاخههای گیاه A را برای تشکیل گروه آزمایشی از گیاه A سمپاشی کنیم و برای تشکیل گروه شاهد، شاخههای دیگر گیاه A را سمپاشی نکنیم.همین کار را برای B و C انجام دهید.
تکثیر فنی (تکثیر فنی): این یک آزمایش تکراری است که برای جلوگیری از خطاهای ناشی از عملیات طراحی شده است، که در واقع یک سوراخ تکراری است که در همان ماده وجود دارد.هر دو تیمار و کنترل باید دارای تنظیمات تکرار شونده (حداقل سه) از ژن هدف و ژن مرجع داخلی باشند.
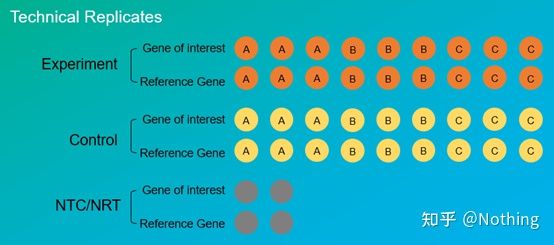
تکرار فنی
فلفل تحت درمان با آفت کش ها را دوباره به عنوان مثال در نظر بگیرید.برای گروه آزمایشی گیاه A، سه سوراخ PCR 1، 2 و 3 به ترتیب برای ژن هدف و ژن مرجع داخلی آن ایجاد کردیم تا میانگین پس از تشخیص بدست آید.برای کنترل گیاه A، گروههای A نیز به همین روش درمان میشوند.به طور مشابه، درمان مشابهی را برای گیاهان B و C انجام دهید.این تکرار فنی است.
شایان ذکر است کهآنچه وارد آمار می شود تکرار بیولوژیکی است و تکرار فنی برای آزمایش این است که آیا پدیده های تصادفی در فرآیند آزمایش وجود دارد یا خیر تا نتایج آزمایشی معتبر باشد، یعنی با گرفتن میانگین آنها همانطور که اغلب می گوییم از خطا جلوگیری شود.
کنترل های منفی - NTC و NRT
NTC (کنترل بدون الگو)، یک کنترل بدون الگو، برای تأیید اینکه آیا مواد آزمایشی آلوده هستند استفاده می شود.معمولاً از آب به عنوان الگو استفاده می شود.اگر یک واکنش فلورسنت وجود داشته باشد، نشان می دهد که آلودگی اسید نوکلئیک در آزمایشگاه رخ داده است.
این آلودگیها از موارد زیر ناشی میشوند: آب ناخالص، معرفهای غیرمجاز حاوی DNA درونزا، آلودگی پرایمر، آلودگی تجهیزات آزمایشگاهی، آلودگی آئروسل و غیره، نیاز به استفاده از روبندههای RNase و مهارکنندههای RNase دارند.پیدا کردن آلودگی آئروسل سخت ترین است.تصور کنید آزمایشگاه شما مانند دود است، با اسیدهای نوکلئیک مختلف در هوا معلق است.

NRT (بدون رونوشت معکوس)، کنترل بدون رونویسی معکوس، RNA رونویسی نشده معکوس به عنوان یک کنترل منفی است که کنترل باقیمانده gDNA است.
هنگام انجام بیان ژن، مقدار RNA با تشخیص مقدار cDNA پس از رونویسی معکوس شناسایی می شود.اگر هنگام خالص سازی RNA باقیمانده gDNA وجود داشته باشد، باعث ایجاد خطا در نتایج تجربی می شود، زیرا نتایج واقعی به دست آمده gDNA و cDNA هستند.در سطح کل، نه فقط cDNA، gDNA باید به طور کامل در طول استخراج RNA حذف شود.
MIQE (2) - اطلاعات نمونه
به اصطلاح اطلاعات نمونه به این معنی است که وقتی مقاله ای در مورد qPCR منتشر می کنیم، باید اطلاعات نمونه را به وضوح توضیح دهیم، که بخشی ضروری از مقاله است.به همین ترتیب، زمانی که نمونهها را پردازش میکنیم، باید عملیات خود را نیز تنظیم کنیم تا از اعتبار نمونهها اطمینان حاصل کنیم.

شرح نمونه فقط یک نتیجه است و باید در کل آزمایش به مواد گرفته شده توجه بیشتری داشته باشیم.
انتخاب مواد آزمایشی
نمونه خون - خون تازه را انتخاب کنید، حداکثر 4 ساعت.نمونه های سلولی - انتخاب کنید تا سلول های تازه را در یک دوره رشد شدید جمع آوری کنید.بافت حیوانی - بافت تازه و با رشد قوی را انتخاب کنید.بافت گیاهی – بافت تازه و جوان را انتخاب کنید.
حتما متوجه شده اید که در این چند جمله یک کلمه کلیدی وجود دارد: تازه.
برای نمونه های فوق، بهترین، مقرون به صرفه و کیت پایدار موجود در بازار، کیت Foregene است که می تواند به سرعت و به راحتی DNA و RNA آنها را استخراج کند.
Plant Total RNA Isolation Kit Plus
ذخیره سازی مواد آزمایشی
به طور کلی، اگر شرایط اجازه می دهد، ما ذخیره نمونه ها را توصیه نمی کنیم.با این حال، بسیاری از دوستان هستند که نمی توانند بلافاصله پس از نمونه برداری آزمایش انجام دهند و حتی برخی نیاز به حمل مخازن نیتروژن مایع به مزرعه برای نمونه برداری دارند.
برای این جور دوست سخت کوش فقط میتونم بگم مواد مصرفی معرف رو متوجه نمیشید.اکنون بسیاری از شرکتهای مصرفی معرف، معرفهایی تولید میکنند که میتوانند نمونههای RNA را در دمای اتاق ذخیره کنند و شما میتوانید از آنها استفاده کنید.روش متداول ذخیره سازی، ذخیره نیتروژن مایع است، با استفاده از یک مخزن نیتروژن مایع کوچک که حمل آن آسان است.پس از بازگرداندن نمونه به آزمایشگاه، آن را در یخچال -80 درجه سانتیگراد نگهداری کنید.

برای آزمایشهای مربوط به RNA، اصل شش کلمهای باید رعایت شود:دمای پایین، بدون آنزیم،وسریع .
درک مفهوم دمای پایین آسان است.بدون آنزیم، RNase در همه جای دنیا که ما در آن زندگی می کنیم وجود دارد (در غیر این صورت شما توسط HIV کشته می شدید)، بنابراین نحوه اجتناب از RNase هنگام انجام آزمایش یک مفهوم بسیار مهم است.سریع،هیچ کونگ فوی در دنیا وجود ندارد که نتوان آن را شکست، فقط سرعت را نمی توان شکست.
بنابراین، به یک معنا، هر چه زمان استخراج کوتاهتر باشد، کیت بهتر است.چرافورجینکیت بر سرعت تاکید دارد، زیرا آنها آن را به خوبی می دانند.
پ.ن: بعضی از دختران با دقت زیادی آزمایش می کنند، اما بعد از چندین سال کار به خوبی یک اسلم دانک نیستند.آنها احساس می کنند که خدا بی انصاف است، از دیگران شکایت می کند و به دنبال زندگی است.در واقع، او آن را درک نکرد.او به خوبی از RNA محافظت نکرد و بازیکن اسلم دانک زیرک بود.وقتی آزمایش را انجام می داد، فکر می کرد که اسلم دانک را با سه بار، پنج بار و دو تقسیم به پایان می رساند، اما آزمایش را به خوبی انجام داد.
توجه داشته باشید: آهسته تر، احتمال بیشتر تهاجم RNase.چگونه خود را برای سریع بودن تربیت کنیم؟راهی نیست فقط بیشتر تمرین کنید.
برای آزمایشهای مختلف و نمونههای مختلف، همچنان لازم است ادبیات بیشتری مطالعه شود و روش مناسب برای پردازش انتخاب شود.برای فرآیند جمعآوری و ذخیرهسازی نمونه، MIQE مستلزم آن است که باید به وضوح در مقاله نوشته شود، تا داوران بتوانند قابلیت اطمینان مقاله را بررسی کنند، و همچنین برای جوانان مبهوت شده راحت است که آزمایش شما را تکرار کنند.
اگرچه آزمایشهای بیولوژیکی دشوار هستند، اما سطح بالایی دارند.اگر مراقب نباشید، می توانید دنیا را زیر و رو کنید.برای مثال، تبدیل سارس به یک بحران بیوشیمیایی، یا ساخت برنج هیبریدی برای نجات 1.3 میلیارد نفر.تصویر زیر یک آزمایش شیمیایی است، فقط با نگاه کردن به ظاهر دیک مانند او باید بفهمید که چقدر به تحقیقات خود افتخار می کنید.فراموشش کن، سیاهش نکن.

MIQE (3) - استخراج اسید نوکلئیک.
استخراج اسید نوکلئیک یک رویداد بزرگ است و تمام آزمایشات زیست شناسی مولکولی با استخراج اسید نوکلئیک شروع می شود.اول از همه، بیایید محتوای MIQE در استخراج اسید نوکلئیک را کپی کنیم.
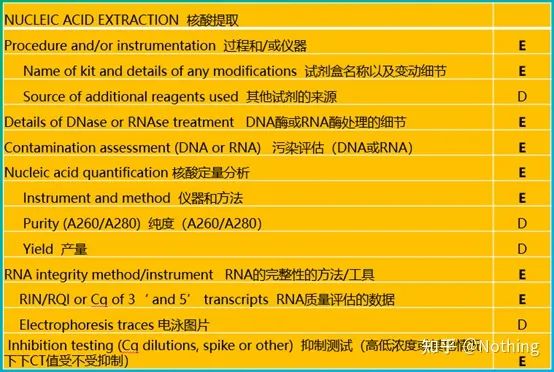
با نگاه کردن به این فرم، نمی توانید روی سطح بمانید.شکل یک دگم است.برای اینکه یک دانش آموز برتر باشید، باید دلیل آن را بپرسید.محتوای ضروری این جدول عبارت است از: پیگیریخلوص، یکپارچگی، قوام و مقدار استخراج RNA .
قسمت اول ازفرآیند یا ابزار مرحله استخراج اسید نوکلئیک است.اگر از دستگاه استخراج خودکار اسید نوکلئیک برای استخراج استفاده می کنید (پیشرفته، لطفاً برای خرید با من تماس بگیرید)، باید نام مدل دستگاه را ذکر کنید.

نام کیت و
چه کیتی برای جزئیات تغییر استفاده شده است، چه معرف های خاصی اضافه شده است یا چه عملیات ویژه ای انجام شده است باید به وضوح توضیح داده شود تا دیگران بتوانند به راحتی آزمایش شما را تکرار کنند.
برخی از افراد هنگام استخراج نمونه های خاص، به این فکر می کنند که این اسلحه مخفی آنهاست و به دیگران نمی گویند، معرف های خاصی را اضافه می کنند.در حالی که آن را مخفی نگه می دارند، فرصت درخشش مقاله شما را نیز از دست می دهند.باهوش نباشید، باید در تحقیقات علمی از ژانگ پیر کشور صادق تر باشید، اگر می خواهید باهوش باشید، مقاله شما را احمق می کند.
باید شماره محصول کیت را به خاطر بسپاریدوقتی کیت را سفارش می دهید و مقاله را می نویسید.به طور کلی دو شماره روی کیت وجود دارد: Cat-شماره کاتالوگ (شماره محصول، شماره مقاله)، Lot-شماره تعداد محصول (برای نشان دادن اینکه محصول از کدام دسته تولید شده است استفاده می شود).
علاوه بر این، شماره CAS اغلب هنگام سفارش معرف های بیوشیمیایی استفاده می شود و من آن را با هم محبوب خواهم کرد.شماره CAS عددی است که انجمن شیمی آمریکا به هر داروی شیمیایی جدید داده است.به طور کلی، سه عدد با یک خط تیره به هم متصل می شوند.شماره CAS Ruhui: 7732-18-5.مواد شیمیایی اغلب چندین نام مستعار دارند، اما شماره CAS منحصر به فرد است.هنگام سفارش دارو، می توانید ابتدا شماره CAS آن را بررسی کنید.
نزدیکتر به خانه، چرا باید این موارد را به وضوح توصیف کنیم؟در واقع، این نیز برای بررسی کیفیت استخراج RNA است.استفاده از ابزارها و کیت ها استخراج RNA را سازگارتر می کند.مقیاس استخراج آزمایشگاه های معمولی زیاد نیست و با کیت می توان آن را به دست آورد.
جزئیات درمان DNase یا RNase
مسئله مهم PCR کمی فلورسنت جلوگیری از آلودگی DNA است و در صورت وجود آلودگی آزمایش نکنید.بنابراین، ضروری است که فرآیندی را که برای پردازش DNA استفاده کردید، بیان کنید تا نشان دهید که DNA در فرآیند آزمایشی به طور کامل و کامل حذف شده است.توسط یک نمودار شماتیک نشان داده شده است.
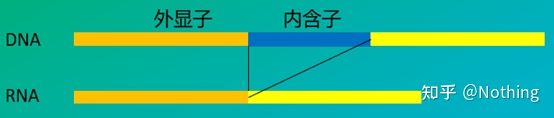
نمودار شماتیک RNA و DNA
به طور کلی، روش حذف DNA، تیمار RNA با DNase پس از استخراج است.با این حال، این روش های نسبتا قدیمی هستند.کیت های استخراج RNA تجاری قادر به حذف DNA در طول فرآیند استخراج بدون افزودن DNase بوده اند.به عنوان مثال، یک سری کیت از Foregene.
توجه داشته باشید: حذف DNA در حین استخراج RNA یک شمشیر دولبه بسیار خطرناک است که زمان عملیات استخراج RNA را طولانی کرده و خطر تخریب RNA را افزایش می دهد.اساساً، این یک مبادله بین بازده و خلوص RNA است.
علاوه بر این، مقدار DNase اضافه شده به ستون جذب مبتنی بر سیلیس بسیار کم است و برای رسیدن به اثر باید از DNase با کیفیت بالا استفاده شود.DNase بهینه نشده را نمی توان به سرعت و به طور کامل هضم کرد.این تست سطح فنی تاجر است.البته، بازرگانان عجیبتری هم وجود دارند که به خود میبالند که DNA را میتوان بدون DNase حذف کرد.می توان گفت هر کسی که لاف می زند که DNA را می توان به طور کامل بدون DNase حذف کرد، یک هولیگان است.DNA یک ساختار دو رشته ای نسبتاً پایدار است و فقط با صحبت کردن و خندیدن نمی توان آن را از بین برد.
ارزیابی آلودگی
روش ارزیابی: تشخیص الکتروفورز، آگارز 1٪، 6V/cm، 15 دقیقه، بارگذاری 1-3 ul
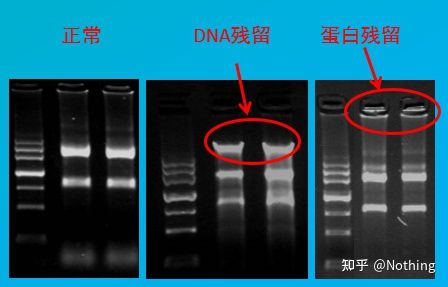
تجزیه و تحلیل کمی اسید نوکلئیک
معمولاً با استفاده از اسپکتروفتومتر UV اندازه گیری می شود.اجازه دهید ابتدا معنای سه مقدار OD260، OD280 و OD230 را رایج کنم.
·OD260nm: طول موج جذب بالاترین پیک جذب اسید نوکلئیک است و بهترین مقدار اندازه گیری شده بین 0.1 تا 1.0 است.در غیر این صورت، نمونه را رقیق یا غلیظ کنید تا در محدوده قرار گیرد.
·OD280nm: طول موج جذب بالاترین پیک جذب پروتئین و مواد فنلی است.
·OD230nm: طول موج جذب بالاترین پیک جذب کربوهیدرات ها است.
بعد، بیایید در مورد نقش هر شاخص صحبت کنیم.برای A260 می توان از آن برای اندازه گیری بازده اسید نوکلئیک استفاده کرد.وقتی OD260=1، dsDNA=50μg/ml، ssDNA=37μg/ml، RNA=40μg/ml.
برای خلوص، باید به نسبت هایی که معمولاً می بینیم نگاه کنیم: OD260/280 و OD260/230.
·DNA خالص: OD260/280 تقریباً برابر با 1.8 است.وقتی بیشتر از 1.9 باشد، نشان دهنده وجود آلودگی RNA و زمانی که کمتر از 1.6 باشد، نشان دهنده وجود آلودگی پروتئین و فنل است.
·RNA خالص: 1.7
·OD260/230: چه DNA باشد چه RNA، مقدار مرجع 2.5 است.وقتی کمتر از 2.0 باشد، نشان دهنده وجود آلودگی قند، نمک و مواد آلی است.
یکپارچگی RNA
اندازه گیری یکپارچگی RNA بسیار مهم است.به طور کلی، لازم است یک آزمایش ژل دناتوراسیون RNA انجام شود تا بررسی شود که آیا روشنایی بین RNA 28S و 18S یک رابطه دو برابری است یا خیر.هنگامی که باند سوم 5S ظاهر می شود، به این معنی است که RNA به جز بی مهرگان شروع به تجزیه کرده است.

دادههای ارزیابی کیفیت RNA: علاوه بر آزمایشهای فوق، آزمایشهای ابزار پیشرفتهتری نیز از نظر یکپارچگی RNA وجود دارد، مانند آزمایش یکپارچگی RQI سیستم الکتروفورز خودکار Experion که میتواند تشخیص دهد که آیا RNA به طور نامرئی تجزیه میشود یا خیر.
در تحقیقات علمی، PCR کمی فلورسنت مقایسه بین ژن هدف و ژن مرجع داخلی است.بنابراین، در فرآیند نگهداری نمونه RNA، استخراج RNA و غیره، هدف اولیه اطمینان از یکپارچگی RNA است.
اینکه چگونه یکپارچگی RNA بر تعادل بین ژن هدف و ژن مرجع داخلی تأثیر می گذارد، می توان به راحتی از شکل زیر فهمید.تخریب منجر به ناقص بودن ژن خواهد شد، چه ناقص بودن ژن مرجع داخلی و چه ناقص بودن ژن هدف، تاثیر زیادی بر روی داده ها خواهد داشت.

نمودار شماتیک ژن هدف و ژن مرجع، نباید درست باشد
تست بازداری (این که آیا مقدار CT تحت غلظت بالا یا پایین یا شرایط دیگر سرکوب می شود)
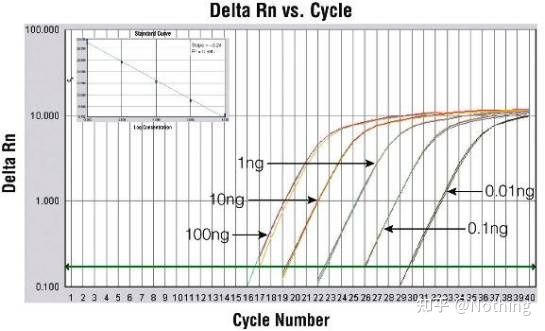
با در نظر گرفتن این شکل به عنوان مثال، مقادیر Ct پنج منحنی به شرح زیر است.توزیع مقادیر CT بین منحنی ها ناهموار است و مقادیر Ct در غلظت های بالا و پایین به تأخیر می افتد که در مورد مهار PCR است.
نکته کلیدی: در فرآیند استخراج RNA، باید باورهای غلط را کنار بگذاریم و باورهای صحیح را ایجاد کنیم.
ایده اشتباه این است: استخراج RNA فقط به دنبال بازده است، با این تصور که هر چه مقدار RNA به دست آمده بیشتر باشد، بهتر است.در واقع، وقتی کمیت را انجام می دهیم، اگر تعداد ژن ها خیلی زیاد نباشد، به RNA زیادی نیاز نداریم.مقدار RNAی که استخراج می کنید بیش از اندازه کافی است.
مفهوم صحیح این است:استخراج RNA باید خلوص، یکپارچگی و سازگاری را دنبال کند.خلوص می تواند تضمین کند که رونویسی معکوس بعدی مهار نمی شود و داده ها تحت تأثیر DNA قرار نمی گیرند.یکپارچگی تعادل توالی های هدف و مراجع داخلی را تضمین می کند.سازگاری بارگذاری پایدار نمونه را تضمین می کند.
MIQE (4) - رونویسی معکوس
تصور غلط: پیگیری حجم نمونه بالاتر.
مفهوم درست: به دنبال سازگاری (پایداری)، صرف نظر از مقدار RNA بارگیری شده، کارایی رونویسی معکوس ثابت می ماند و اطمینان حاصل می کند که تفاوت در cDNA می تواند واقعاً منعکس کننده تفاوت در mRNA باشد.
ما این فرآیند را با یک نمودار شماتیک توضیح می دهیم:
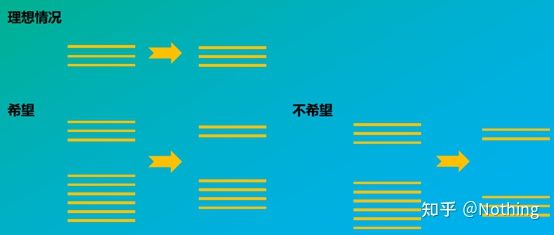
نمودار شماتیک بازده رونویسی معکوس، درست نیست
اول از همه، ما باید تفاوت بین فرآیند رونویسی معکوس و فرآیند PCR را درک کنیم.PCR تحت چندین فرآیند حرارت دهی و بازپخت قرار می گیرد و قطعه هدف به طور تصاعدی رشد می کند.در حالی که رونویسی معکوس این فرآیند را ندارد، می توانیم تصور کنیم که رونویسی معکوس در واقع یک به یک است در طول فرآیند همانندسازی، به تعداد قطعات RNA
از آنجایی که میتوان تعداد زیادی از اطلاعات cDNA را دریافت کرد، باید تا به حال فهمیده شود، زیرا قطعات بزرگ و کوچک رونویسی معکوس شدهاند و تمرکز بر یک قطعه غیرممکن است.و از آنجایی که مقدار RNA نسبتاً کم است، مقدار cDNA بدست آمده نیز نسبتاً کم است، برخلاف PCR که دارای اثر تقویتی است، بنابراین اساساً تشخیص آن غیرممکن است.
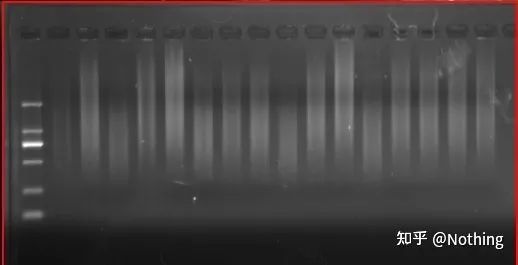
نتایج الکتروفورز cDNA
دوم اینکه، در حالت ایده آل، رونویسی معکوس یک به یک انجام می شود، اما هیچ رونویسی معکوس از هیچ شرکتی نمی تواند به این اثر دست یابد.اساسا، کارایی بیشتر ترانس کریپتازهای معکوس بین 30 تا 50 درصد است.اگر اینطور باشد، ما ترجیح میدهیم بازده رونویسی معکوس نسبتاً پایداری داشته باشیم، این همان چیزی است که میخواهیم در شکل ببینیم: 3 RNA 2 cDNA دریافت میکنند، 6 RNA 4 cDNA دریافت میکنند، بنابراین مهم نیست که چقدر نمونه بارگذاری شده است، بازده رونویسی معکوس نسبتاً پایدار است.ما نمیخواهیم شرایطی را ببینیم که بازده رونویسی معکوس ناپایدار است و غلظت بالا مهار میشود.
بنابراین، چگونه می توان بررسی کرد که آیا بازده رونویسی معکوس پایدار است؟روش بسیار ساده است، فقط باید یک آزمایش مقایسه انجام دهید: یکی این است که پس از رقیق شدن دوبرابر RNA رونویسی معکوس به cDNA انجام دهید، و دیگری این است که پس از رونویسی معکوس در cDNA، رقت دو برابری انجام دهید و سپس qPCR را انجام دهید تا شیب به دست آمده را ببینید آیا سازگار است.به عنوان یک دانش آموز برتر، باید آن را در چند ثانیه درک کنید.همانطور که در زیر نشان داده شده است:
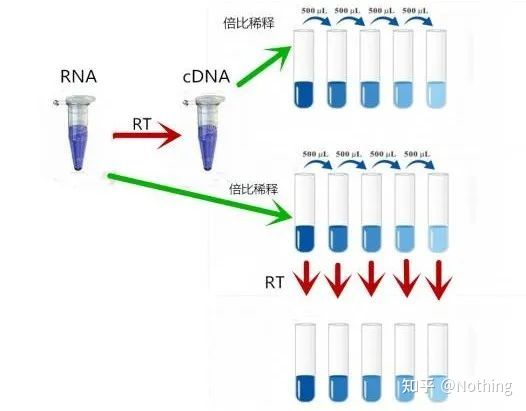
رقیق کردن RNA و cDNA برای آزمایش اینکه آیا کارایی رونویسی معکوس پایدار است یا خیر
رونوشت معکوس و کیت
چگونه می توان PCR کمی فلورسنت کامل دارای رونوشت و کیت معکوس عالی باشد.ترانس کریپتاز معکوس به طور کلی بر اساس منبع به دو نوع تقسیم می شود، AMV یاM-MLVو عملکرد آنها مانند جدول نشان داده شده است.
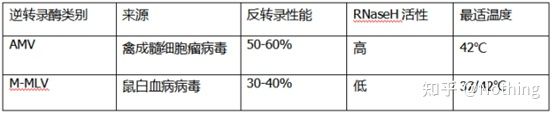
فعالیت RNase H
RNase H ریبونوکلئاز H است، نام چینی ریبونوکلئاز H است که یک اندوریبونوکلئاز است که می تواند به طور خاص RNA را در زنجیره هیبریدی DNA-RNA هیدرولیز کند.RNase H نمی تواند پیوندهای فسفودی استر را در DNA یا RNA تک رشته ای یا دو رشته ای هیدرولیز کند، یعنی نمی تواند DNA یا RNA تک رشته ای یا دو رشته ای را هضم کند.معمولاً در سنتز رشته دوم cDNA استفاده می شود.
چیز عجیبی است.ما می گوییم که ترانس کریپتاز معکوس دارای فعالیت RNase H است، نه اینکه ترانس کریپتاز معکوس حاوی RNase H است و ممکن است جدا کردن RNase H از ترانس کریپتاز معکوس امکان پذیر نباشد، شاید به دلیل ترکیب گروه های خاصی در ترانس کریپتاز معکوس این فعالیت توسط ترانس کریپتاز معکوس ایجاد می شود.
بنابراین، صرف نظر از کارایی بالاتر رونویسی معکوس AMV، فعالیت RNase H آن باعث کاهش بازده cDNA می شود.البته، تولیدکنندگان معرف به طور مداوم محصولات خود را بهینه سازی می کنند تا فعالیت RNase H در رونوشت معکوس را تا حد امکان حذف کنند تا بازده cDNA را افزایش دهند.
دمای بازپخت
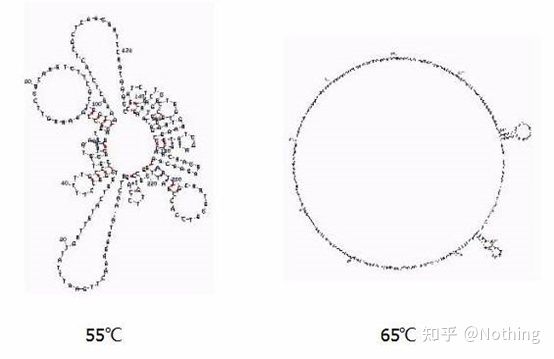
ساختار ثانویه RNA در دماهای مختلف
شکل بالا را برای ساختار ثانویه RNA در دماهای مختلف ببینید و از ابزار آنلاین mFold برای تعیین ساختار ثانویه قطعه هدف در شرایط دمایی و غلظت نمک خاص استفاده کنید.در دمای 55 درجه سانتیگراد، ساختار ثانویه RNA هنوز بسیار پیچیده است، ترانس کریپتاز معکوس نمی تواند کار کند، و ساختار ثانویه تا دمای 65 درجه سانتیگراد نمی تواند به طور کامل حل شود، در حالی که دمای بهینه AMV و M-MLV بسیار کمتر از این دما است.
چه باید کردساختار ثانویه جفت شدن مکمل خود الگو است که منجر به رقابت شدید بین پرایمر و رونوشت معکوس و الگو می شود و در نتیجه یک سری مشکلات مانند E پایین و تکرارپذیری ضعیف ایجاد می شود.
چه باید کردفقط دمای بازپخت را تا حد امکان افزایش دهید.
بسیاری از تولیدکنندگان معرف در حال بهبود رونوشت معکوس خود از طریق مهندسی ژنتیک هستند.برخی دمای واکنش را افزایش می دهند، مانند جیفان و آیدلای، و برخی گروه فعال آنزیم RNase H را حذف می کنند تا میل ترکیبی بین آنزیم و الگوی RNA را بهبود بخشند.میل ترکیبی بالا می تواند ساختار ثانویه را به صورت رقابتی فشرده کند و به آرامی بخواند و همچنین کارایی رونویسی معکوس را تا حد زیادی بهبود بخشد.
نکته کلیدی: رونویسی معکوس برای پیگیری سازگاری بازده رونویسی معکوس مهمتر است (آنزیم ها نه تنها باید کارآمد باشند بلکه باید پایدار باشند)، به جای مقدار نمونه بارگذاری شده، اگر PCR کمی فلورسنت در مقیاس بزرگ نباشد، به هیچ وجه امکان پذیر نخواهد بود.cDNA های متعدد
تولید کنندگان مختلف نیز تلاش هایی را در جهت دستیابی به ثبات انجام داده اند.به عنوان مثال، اکنون اکثر شرکت ها رونویسی معکوس را به عنوان یک کیت استاندارد برای فروش بسته بندی کرده اند که انتخاب خوبی است.
به عنوان مثال، کیت های سری RT Easy Foregene:
RT Easy I (پرمیکس اصلی برای کیت سنتز cDNA رشته اول)
MIQE (5) - اطلاعات ژن هدف

شکل بالا توضیح می دهد
1. اینکه آیا این ژن برای آزمایش های مکرر مؤثر است یا خیر، عموماً می توان با آزمایش های مکرر تأیید کرد.
2. شناسه ژن، می دانید.
3. طول ژن، طول کل ژن هدف قطعا مشکلی ندارد.هنگام طراحی پرایمرها، اطمینان حاصل کنید که طول آمپلیکون بین 80-200 جفت باز باشد تا از راندمان تقویت بهتر اطمینان حاصل شود.
4. اطلاعات مقایسه Sequence Blast، ژن هدف باید در بانک ژن مقایسه شود تا از تقویت غیر اختصاصی جلوگیری شود.
5. وجود شبه زا.کاذب یک توالی DNA شبیه به یک ژن طبیعی است اما عملکرد طبیعی خود را از دست می دهد.اغلب در خانواده چند ژنی یوکاریوت ها وجود دارد.معمولاً با ψ نشان داده می شود.این یک کپی DNA ژنومی غیرعملکردی در ژنوم است که بسیار شبیه به توالی ژن کد کننده است.، عموماً رونویسی نمی شوند و معنای فیزیولوژیکی روشنی ندارند.
6. موقعیت آغازگرها نسبت به اگزون ها و اینترون ها.در سالهای اولیه، زمانی که مشکل آلودگی DNA را حل کردیم، اغلب به موقعیت آغازگرها، اگزونها و اینترونها توجه میکردیم و عموماً طراحی پرایمرها را در طول اینترونها برای جلوگیری از تقویت DNA در نظر میگرفتیم.لطفاً شکل زیر را ببینید: سیاه نشان دهنده اینترون ها، آبی های مختلف نشان دهنده اگزون ها، صورتی نشان دهنده آغازگرهای رایج، و قرمز روشن نشان دهنده آغازگرهای پوشاننده اینترون هستند.
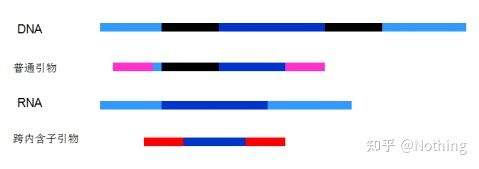
شماتیک، هرگز درست نیست
چه طرح کاملی به نظر می رسد، اما در واقع، در بیشتر موارد، پرایمرهای ترانس اینترون به اندازه تصور جادویی نیستند و همچنین باعث تقویت غیر اختصاصی می شوند.بنابراین بهترین راه برای جلوگیری از آلودگی DNA حذف کامل DNA است.
7. پیش بینی ساختار.با استفاده مجدد از این مثال، از ابزار آنلاین mFold برای تعیین ساختار ثانویه قطعه هدف در دما و غلظت نمک خاص استفاده کنید.
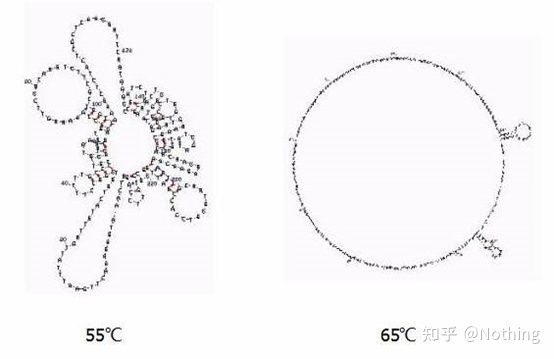
ساختار ثانویه RNA در دماهای مختلف
ساختار ثانویه جفت شدن مکمل خود قالب است که منجر به رقابت شدید بین جفت شدن پرایمر و الگو خواهد شد و احتمال اتصال پرایمر کمتر است و در نتیجه یک سری مشکلات مانند E پایین و تکرارپذیری ضعیف ایجاد می شود.از طریق پیش بینی نرم افزار، اگر مشکل ساختار ثانویه وجود نداشته باشد، عالی خواهد بود.اگر وجود داشته باشد، مقاله بعدی ما به طور خاص نحوه حل این مشکل را مورد بحث قرار خواهد داد.
MIQE (6) - الیگونوکلئوتیدهای qPCR

برای PCR کمی فلورسنت، اولین چیزی که هر روز با آن دست و پنجه نرم می کنید استخراج RNA است و دومین مورد ممکن است طراحی پرایمر باشد.
اول از همه، ما همچنان قوانین مربوط به طراحی پرایمر را طبق چک لیست MIQE بررسی می کنیم.به قدری ساده است که آشغال ها می توانند بخندند و ما می توانیم آن را در یک جمله تمام کنیم: دنباله و موقعیت کاوشگر پرایمر و روش اصلاح را بیابید.برای روش خالصسازی پرایمر، سنتز پرایمر در حال حاضر بسیار ارزان است، qPCR شایسته روشهای خالصسازی PAGE و بالاتر است و اطلاعات ابزار سنتز مهم نیست.بسیاری از مردم چندین دهه است که پرایمر انجام می دهند و نمی دانند که سینت سایزر ABI3900 است.
با توجه به اصول طراحی پرایمر، لازم نیست آنها را به طور خلاصه به خاطر بسپارید، زیرا اکثر نرم افزارهای طراحی پرایمر یا ابزارهای آنلاین می توانند این مشکلات را برطرف کنند (ابزار آنلاین توصیه شده primer3.ut.ee/) و 99.999% طراحی پرایمر به صورت دستی انجام نمی شود.
فقط نکات زیر را بعد از طراحی پرایمرها بررسی کنید:
1. طراحی پرایمرهای نزدیک به انتهای 3: در صورت استفاده از پرایمرهای الیگو dT برای سنتز رشته اول cDNA، با توجه به کارایی رونویسی معکوس و یکپارچگی RNA، پرایمرهای طراحی شده باید نزدیک به انتهای 3 طراحی شوند تا راندمان تقویت افزایش یابد.از یک تصویر برای توضیح به صورت زیر استفاده کنید (هیچ راهی برای درک این موضوع وجود ندارد):
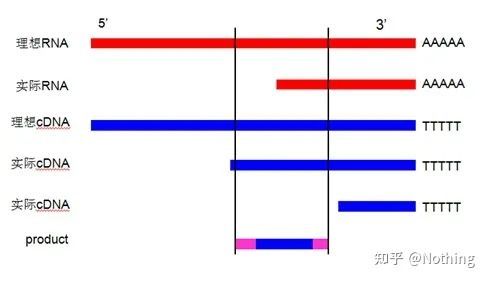
چرا باید پرایمرها نزدیک به انتهای 3 طراحی شوند، نباید درست باشد
2. مقدار TM: مقدار Tm در 55-65 درجه سانتیگراد است (زیرا فعالیت اگزونوکلئاز در دمای 60 درجه سانتیگراد بالاترین است) و محتوای GC در 40٪ -60٪ است.
3. BLAST: برای جلوگیری از تکثیر غیر اختصاصی ژنوم، Blast باید برای تایید تکمیلی استفاده شود.
MIQE (7) - فرآیند qPCR
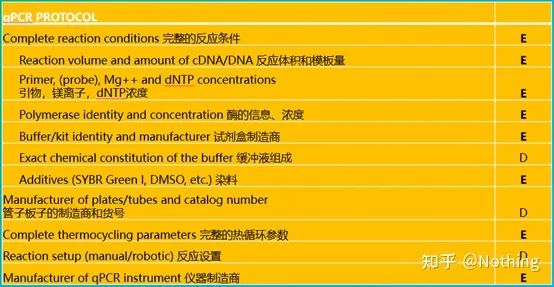
1. کیت qPCR
با توجه به الزامات MIQE، ما باید شرایط واکنش کامل را در مقاله به وضوح شرح دهیم، از جمله پیکربندی سیستم واکنش PCR، کیت مورد استفاده، سازنده کیست، اندازه سیستم واکنش چقدر است، روش رنگ یا پروب استفاده شده است، تنظیمات برنامه PCR.رانندگان کهنه کار قطعا متوجه خواهند شد که تا زمانی که کیت انتخاب شده باشد، اساساً اطلاعات فوق مشخص می شود.
در حال حاضر، ساخت و تولید کیت های فلورسنت کمی PCR یک فناوری بسیار بالغ است.تا زمانی که تولیدکنندگان بسیار بدی را انتخاب نکنید، احتمال بروز مشکلات زیاد نیست، اما همچنان می خواهیم چند نکته را با شما در میان بگذاریم:
آنزیم Taq شروع داغ:مهمترین بخش PCR آنزیم Taq شروع داغ است.آنزیمهای شروع داغ موجود در بازار به طور کلی به دو نوع تقسیم میشوند، یکی آنزیم استارت گرم اصلاحشده شیمیایی (شما میتوانید آن را به عنوان جاسازی پارافین تصور کنید)، و دیگری آنزیم استارت داغ برای اصلاح آنتیبادی (پیوند آنتیژن به آنتیبادی) است.اصلاح شیمیایی یک راه اولیه برای شروع گرم آنزیم ها است.وقتی به دمای معینی رسید، آنزیم فعالیت خود را آزاد می کند.آنزیم شروع گرم اصلاح شده با آنتی بادی از روش های بیولوژیکی برای جلوگیری از فعالیت آنزیم استفاده می کند.وقتی به دمای معینی رسید، آنتی بادی دناتوره شده و به عنوان پروتئین غیرفعال می شود و فعالیت آنزیم وارد عمل می شود.
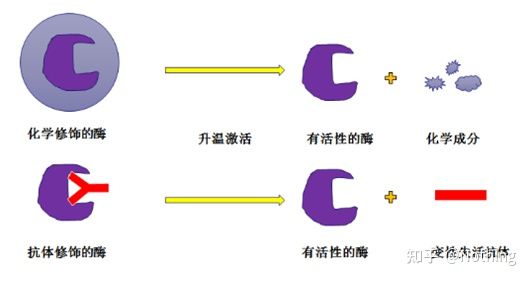
با این حال، این چه فایده ای دارد؟در این صورت، فعالیت آزادسازی آنزیمهای اصلاحشده با آنتیبادی سریعتر از آنزیمهای اصلاحشده شیمیایی است، بنابراین از نظر حساسیت، آنزیمهای اصلاحشده با آنتیبادی یک مزیت جزئی دارند، به طوری که اساساً هیچ آنزیم اصلاحشده شیمیایی در کیتهای موجود در بازار وجود ندارد.اگر وجود داشته باشد، پس فناوری این سازنده هنوز در عصر هزاره گیر کرده است.
غلظت یون منیزیم:غلظت یون منیزیم در واکنش PCR بسیار مهم است.غلظت مناسب یون منیزیم می تواند باعث آزاد شدن فعالیت آنزیم Taq شود.اگر غلظت خیلی کم باشد، فعالیت آنزیم به طور قابل توجهی کاهش می یابد.اگر غلظت بیش از حد بالا باشد، تقویت غیر اختصاصی کاتالیز شده با آنزیم افزایش می یابد.غلظت یونهای منیزیم نیز بر بازپخت پرایمرها، دمای ذوب قالب و محصولات PCR تأثیر میگذارد و در نتیجه بر بازده قطعات تقویتشده تأثیر میگذارد.غلظت یون منیزیم به طور کلی در 25 میلی مولار کنترل می شود.البته برای یک کیت خوب باید غلظت یون منیزیم به خوبی کنترل شود.برخی از بازرگانان یک عامل کیل کننده یون منیزیم را به معرف اضافه می کنند که می تواند به اثر تنظیم خودکار غلظت یون منیزیم دست یابد.
غلظت رنگ فلورسنت:رنگ فلورسنت ، که رنگ سبز Sybr است که ما معمولاً از آنها استفاده می کنیم ، به طور عمده با اتصال به شیار جزئی DNA دو رشته ای ، فلورسانس تولید می کند ، زیرا اتصال رنگ به DNA دو رشته ای غیر خاص است ، یعنی تا زمانی که DNA دو طبقه با آن ترکیب شود ، فلورسانس می تواند رخ دهد ، بنابراین می توان آن را به وجود آورد ، بنابراین رنگ های آغازگر و DNA با استفاده از DNA با استفاده
PS: محصولات موجود در بازار به دلیل خاصیت حساس به نور، عموماً در لوله های سانتریفیوژ مات قهوه ای (مانند تصویر زیر) بسته بندی می شوند.با این حال، این با مشکل مواجه خواهد شد.تشخیص اینکه آیا مایع در هنگام نمونه برداری مکیده می شود یا خیر دشوار است.از این نظر، Qingke در واقع کاربرپسندترین است (همانطور که در تصویر زیر نشان داده شده است)، و لوله شفاف در یک کیسه قلع مات بسته بندی شده است.سپس با در نظر گرفتن راحتی اجتناب از نور و نمونه برداری، آن را در یک کیسه حلبی قرار دهید.شما باید شماره محصول مناسب را انتخاب کنید.TSE204 یک موجود بسیار مقرون به صرفه است که باعث می شود من بخواهم چمن بکارم.

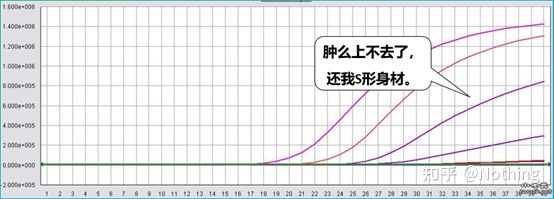
غلظت رنگ فلورسنت نیز بسیار مهم است.اگر غلظت خیلی کم باشد، منحنی تقویت در مرحله بعدی بالا نمی رود و کامل نیست.اگر غلظت بیش از حد بالا باشد، باعث تداخل نویز می شود.از آنجایی که PCR کمی فلورسنت عمدتاً به مقدار CT بستگی دارد، اگر غلظت رنگ فلورسنت به درستی تنظیم نشود، نقطه پایین بهتر از نقطه بالا است.البته غلظت رنگ مناسب بهترین است.
ROX: رنگ های ROX برای تصحیح خطاهای سیگنال فلورسانس به خوبی استفاده می شود.برخی از سازندگان ابزار نیاز به کالیبراسیون دارند، در حالی که برخی دیگر نیازی به کالیبراسیون ندارند.به عنوان مثال، استفاده از ابزار تقویت کننده Real Time PCR شرکت Thermo Fisher Scientific معمولاً نیاز به کالیبراسیون دارد که شامل 7300، 7500، 7500Fast، StepOnePlus و غیره می شود. دستورالعمل های کلی کیت آن را توضیح می دهد.
مخلوط qPCR Foregene همچنین حاوی رنگ ROX است که برای استفاده در مدل های مختلف مناسب است.
درمان پیوند هیدروژنی ضعیف: درمان پیوندهای هیدروژنی ضعیف یک موضوع نسبتاً فنی است.هیچ چیز دفترچه راهنمای بسیاری از کیت ها را نخوانده است، اما هیچ یک از آنها به این موضوع اشاره نکرده اند.در واقع خیلی مهم است.ترکیب بازها عمدتاً به استحکام پیوندهای هیدروژنی بستگی دارد.پیوندهای هیدروژنی قوی تقویت عادی هستند و پیوندهای هیدروژنی ضعیف منجر به تقویت غیر اختصاصی می شوند.اگر پیوندهای هیدروژنی ضعیف را نتوان به خوبی از بین برد، نمی توان از تقویت غیر اختصاصی اجتناب کرد.در محدوده نویسنده، تنها تعداد کمی از شرکت ها متوجه این مشکل شده اند.هنگام خرید کیت می توانید به این موضوع مراجعه کنید که آیا برای کیتی که می خواهید انتخاب کنید راه حلی در این زمینه در نظر گرفته اید یا خیر.
حجم واکنش: سیستم 20-50ul بیشتر مورد استفاده قرار می گیرد و حجم های کوچکتر احتمالا باعث خطا می شود.به طور کلی، دستورالعمل های کیت استفاده از حجم واکنش PCR را توصیه می کند.هوشمند نباشید و از حجم های کمتر برای صرفه جویی در هزینه ها استفاده کنید.هدف از.حجم توصیه شده توسط بازرگانان در واقع تست شده است و ممکن است آنها نتوانند مشکل خطاهای ناشی از حجم کم را حل کنند.
2. سازنده و شماره مقاله صفحه لوله
همه اصل PCR کمی فلورسنت را می دانند.جمع آوری فلورسانس عمدتاً از طریق کلاهک های لوله PCR انجام می شود.در انتخاب مواد مصرفی PCR به دو نکته توجه کنید: انتقال نور خوب و مناسب برای دستگاه.به طور کلی، بردها و لوله های مارک های اصلی خوب هستند، اما باید از نظر سازگاری با دقت انتخاب کنید، در غیر این صورت نمی توانید از ساز استفاده کنید.

4. دانش سطح بالا
اعتبارسنجی MIQE (8) - qPCR
این اولویت اصلی qPCR است!قهرمانان زیادی در اینجا به شن و ماسه افتاده اند.البته این امکان نیز وجود دارد که شما خوش شانس باشید و ژن هایی که مطالعه کرده اید ساده باشند، بنابراین در غار یخی در امتداد باد شناور شده اید.اطلاعات تأیید qPCR برای آزمایش قابلیت اطمینان داده ها در نظر گرفته شده است.ما اطلاعات تأیید لازم را به شرح زیر لیست می کنیم:
1. آزمون اختصاصیت
ویژگی تکثیر ژن هدف با بررسی اینکه آیا تصویر الکتروفورز یک باند است یا خیر، آزمایش میشود.تأیید توالی؛منحنی ذوب برای دیدن اینکه آیا نقشه اوج تک است یا خیر.تایید هضم آنزیمی و روش های دیگر.
در اینجا، ما بر روی t تمرکز می کنیمتجزیه و تحلیل تقویت غیر اختصاصی با استفاده از روش منحنی های ذوب.به طور کلی، زمانی که پرایمرها را طراحی می کنیم، اندازه قطعه محصول باید در محدوده 80-200 جفت باز باشد که باعث می شود دمای ذوب محصول PCR 80-85 درجه سانتیگراد باشد.بنابراین، اگر پیک های متفرقه وجود داشته باشد، باید محصولات تقویت غیر اختصاصی دیگری نیز وجود داشته باشد.اگر پیک زیر 80 درجه سانتیگراد ظاهر شود، به طور کلی به عنوان یک دایمر آغازگر در نظر گرفته می شود.اگر پیک بالاتر از 85 درجه سانتیگراد ظاهر شود، به طور کلی به عنوان آلودگی DNA یا تکثیر غیر اختصاصی قطعات بزرگ در نظر گرفته می شود.
توجه: گاهی اوقات تنها یک پیک در 80 درجه سانتیگراد وجود دارد.در این زمان، این مفهوم باید رعایت شود.این احتمال وجود دارد که نتایج تقویت همه دایمرهای آغازگر باشند.
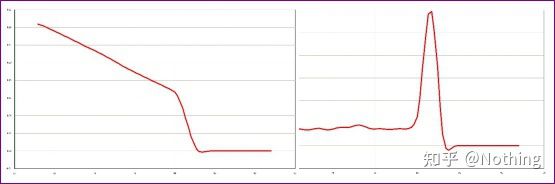
منحنی ذوب نرمال (تک پیک بدون تقویت غیر اختصاصی)
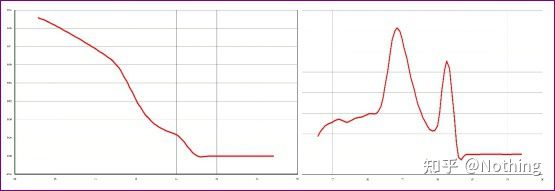
منحنی ذوب مشکل ساز (تقویت غیر اختصاصی پیک های کاذب)
【تحلیل مورد】
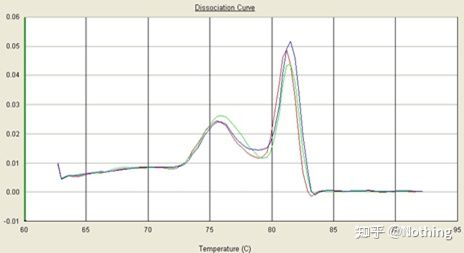
یک قله اصلی وجود دارد، اما دایمر پرایمر جدی است
منحنی ذوب تک قله در شکل زیر می تواند به راحتی چشمان شما را فریب دهد، فکر می کنید این یک آزمایش کامل است، اما نتیجه کاملا اشتباه است.در این زمان، ما باید به دمای ذوب نگاه کنیم.دمای پیک زیر 80 درجه سانتی گراد است که کاملا پرایمر-دایمر است.
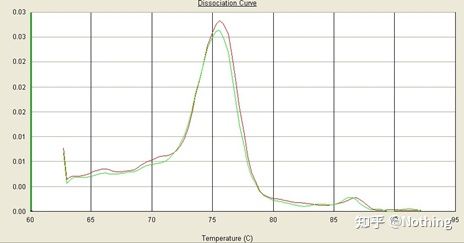
بدون قطعه هدف، تمام دایمرهای آغازگر
اینجا، برادر من نمی تواند متوقف شود.عکس زیر عکسی است که با گوشی موبایل توسط یک آدم کثیف برای من ارسال شده است.معرف هایی که او استفاده می کرد همه مارک های رایج در صنعت هستند.او از یک برند T-prefix به برند T-prefix دیگر تغییر کرد.من فکر می کنم شما قبلا آن را حدس زده اید.آشغال به من گریه کرد: "معرف استفاده شده در تصویر اول خیلی خوب است و اوج آن تک است.بعداً بعد از استفاده از معرف پیشنهادی شما مانند عکس دوم با پیک های مختلط می شود.بدبختم کردی"
دو نمودار را از هم جدا کنید.در نگاه اول یکی دارای قله تکی و دیگری قله دوبل است.مزخرف، یک اوج البته خوب است.آیا این درست است؟
بدتر از Dou E اگه دوتا عکس رو تو عکس زیر بذارم بلافاصله متوجه میشید.در واقع، ما به راحتی با این نوع تصویر فلج می شویم.پس از تجزیه و تحلیل دقیق، متوجه شدیم که: پیک شکل اول در دمای 75 درجه سانتیگراد است که کاملاً دایمر پرایمر است.اوج شکل دوم در 75 درجه سانتیگراد و 82 درجه سانتیگراد ظاهر می شود، حداقل وجود دارد محصول ظاهر می شود.
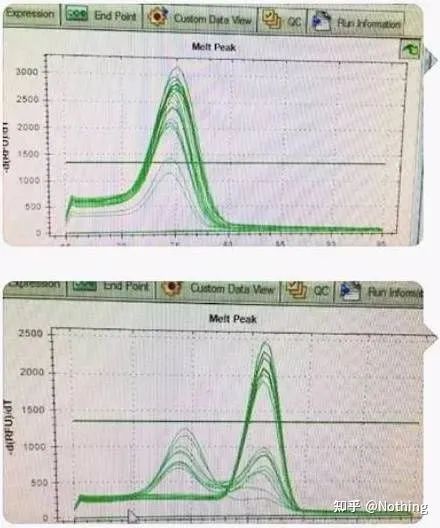
تصاویری از نظرات دانش آموزان
بنابراین مشکل اساسی مشکل معرف ها نیست، بلکه مشکل طراحی پرایمر است.در عین حال ثابت می کند که برخی از مارک های بزرگ از کیفیت آهن برخوردار نیستند و همچنین ثابت می کند که برادرم قبلاً گفته است: این مارک معرف نیست که مقاله شما را پشتیبانی می کند.این مقاله شما است که برند معرف ها را تقویت کرده است.فقط تصور کنید، اگر این آشغال معرف ها را تغییر نمی داد، داده های اشتباه به مجله ارسال می شد و اتفاقی که می افتاد یک تراژدی بود.
2. مقدار Ct کنترل خالی
توضیح ندهید، اگر کنترل خالی مقدار Ct داشته باشد، آلودگی نیست؟با این حال، هنوز باید بدانید که کدام کنترل خالی دارای مقدار Ct است.اگر NTC باشد یعنی DNA خارجی مانند آلودگی معرف وجود دارد.اگر NRT باشد به این معنی است که RNA استخراج شده دارای آلودگی DNA است.
3. منحنی استاندارد
با احتساب شیب و فرمول محاسبه، بازده PCR را می توان از طریق فرمول محاسبه کرد.یک آزمایش کامل نیاز دارد که شیب منحنی استاندارد به 3.32 نزدیک شود و R² به 0.9999 نزدیک شود.
4. محدوده دینامیکی خطی
محدوده دینامیکی واکنش خطی است.با توجه به الگوی مورد استفاده برای تولید منحنی استاندارد، محدوده دینامیکی باید حداقل شامل 5 گرادیان غلظت باشد و به تغییر مقادیر Ct در گرادیان غلظت بالا و گرادیان غلظت پایین توجه شود.
5. دقت تشخیص
تغییرات در نتایج qPCR، یعنی تکرارپذیری ضعیف، یعنی دقت ضعیف، ناشی از عوامل زیادی از جمله دما، غلظت و عملکرد است.دقت qPCR به طور کلی با کاهش تعداد کپی کمتر قابل کنترل می شود.در حالت ایدهآل تنوع درون تجربی، این تنوع فنی باید از تنوع بیولوژیکی متمایز باشد و تکرارهای بیولوژیکی میتوانند مستقیماً تفاوتهای آماری در نتایج qPCR بین گروهها یا تیمارها را بررسی کنند.به ویژه برای سنجش های تشخیصی، بهترین دقت بین سنجش (تکرارپذیری) در بین سایت ها و اپراتورها باید گزارش شود.
6. کارایی تشخیص و LOD (در مولتی پلکس qPCR)
LOD کمترین غلظت 95 درصد از نمونه های مثبت شناسایی شده است.به عبارت دیگر، غلظت LOD موجود در مجموعه ای از تکرارهای ژن هدف نباید از 5% از واکنش های ناموفق تجاوز کند.هنگام انجام آنالیز qPCR مالتی پلکس، به ویژه برای تشخیص همزمان جهشهای نقطهای یا پلیمورفیسمها، qPCR مولتیپلکس باید شواهدی ارائه دهد که دقت قطعات هدف چندگانه در یک لوله به خطر نیفتد، تشخیص چندگانه و تشخیص تک لوله باید یکسان باشد.به خصوص زمانی که ژن های هدف با غلظت بالا و ژن های هدف با غلظت پایین به طور همزمان تقویت می شوند، باید به این مشکل توجه کرد.
مشکلات و راه حل هابه طور کلی، مشکلاتی که اغلب در اشکال زدایی qPCR با آن مواجه می شوند، بر جنبه های زیر تمرکز دارند:
·تقویت غیر اختصاصی
· انتخاب دشوار غلظت پرایمر و مشکل با پرایمر-دایمرها
· دمای بازپخت نادرست است
· ساختار ثانویه بر راندمان تقویت تأثیر می گذارد
تقویت غیر اختصاصی
تقویت غیر اختصاصیاتفاق می افتد، به طور کلی در نظر گرفته می شود که آیا طرح پرایمر مناسب نیست، اما اگر برای تعویض پرایمر عجله ندارید، می توانید ابتدا روش های زیر را امتحان کنید (اصل نیز پیوست شده است):
· افزایش دمای بازپخت - سعی کنید پیوندهای هیدروژنی ضعیفی ایجاد کنید که قادر به حفظ آنها نیستند.
· کوتاه شدن زمان بازپخت و ازدیاد طول - کاهش احتمال پیوندهای هیدروژنی ضعیف.
· کاهش غلظت پرایمر - کاهش احتمال اتصال پرایمرهای اضافی و مناطق غیر هدف.
راندمان تقویت پایین
وضعیت مخالف تقویت غیر اختصاصی - راندمان تقویت پایین و اقدامات برای مقابله با راندمان تقویت پایین دقیقاً برعکس است:
· طولانی شدن زمان بازپخت و ازدیاد طول.
· تغییر به سه مرحله PCR و کاهش دمای بازپخت.
· افزایش غلظت پرایمر.
Ps: بسیاری از دانشجویان فارغ التحصیل متولد دهه 90 تمایلی به مطالعه نحوه اشکال زدایی آزمایشات ندارند و امیدوارند که کیت بتواند مشکل را به طور کامل حل کند (اگر می خواهید بعد از فارغ التحصیلی برای انجام تحقیق و توسعه به یک شرکت معرف مراجعه کنید)، در واقع، سازندگان معرف نیز این گونه فکر می کنند، امیدوارم که احمقانه باشد. عوامل جذب ضعیف پیوند Hبرای حل آسان مشکل، احمق ها هنوز باید مقدمه شرکت معرف را بخوانند تا ببینند آیا عاملی وجود دارد که پیوندهای هیدروژنی ضعیف را جذب می کند یا خیر.
انتخاب دشوار غلظت پرایمر و مشکل با پرایمر-دایمرها
روش 1: به طور کلی، دستورالعمل های کیت برای qPCR دارای سیستم های توصیه شده و غلظت های پرایمر توصیه شده است.
روش 2: اشکال زدایی با تنظیم گرادیان غلظت پرایمر.تصویر زیر برای نشان دادن از یک شرکت به سرقت رفته است.شکل زیر نتایج کمی فلورسانس ساخته شده با سه گرادیان غلظت پرایمر (100nM، 250nM، 500nM) و چهار گرادیان غلظت قالب (0.1ng، 1ng، 10ng، 100ng) را نشان میدهد.مقدار Ct نتایج تجربی به صورت زیر رسم می شود:
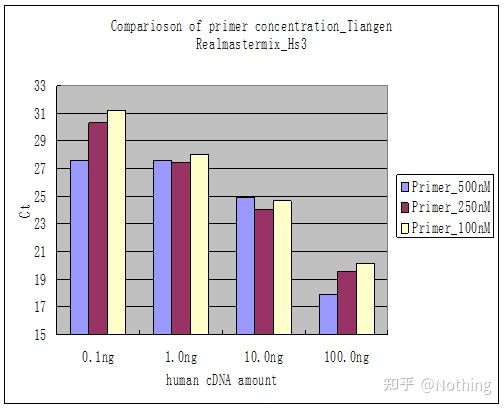
انتخاب غلظت پرایمر هر غلظت آغازگر را به صورت زیر در یک خط الحاق کنید:
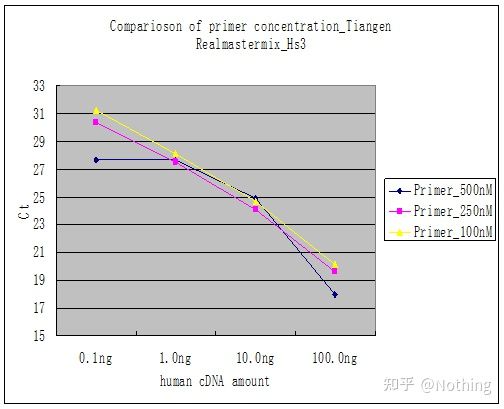
انتخاب غلظت پرایمر واضح است، رابطه خطی غلظت پرایمر 100nM و 250nM بهتر است و رابطه خطی غلظت پرایمر 500nM نسبتا ضعیف است.در 100nM و 250nM، مقدار Ct 250nM نسبتا کم است، بنابراین غلظت بهینه آغازگر 250nM است.به طور کلی پرایمر-دایمرهای شدید را می توان در منحنی ذوب مشاهده کرد.اگر پرایمرهای طراحی شده نتوانند از پرایمر-دایمر اجتناب کنند چه؟
روش 3: مقدار پرایمرها را کاهش دهید و دمای آنیلینگ را افزایش دهید (نیاز به توضیح نیست).
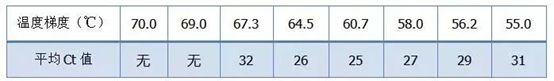
مقدار تجربی دمای بازپخت 60 درجه سانتیگراد است.اگر مطمئن نیستید، چگونه دمای بازپخت مناسب تری را انتخاب کنید؟پاسخ همان انتخاب غلظت آغازگر است -تست گرادیان.برای نشان دادن مشکل از شرکت Bio-rad عکس بگیرید.برای تقویت یک قطعه هدف معین، هشت گرادیان دما را هر کدام با سه تکرار تنظیم کنید و منحنی تقویت به دست آمده به صورت زیر است:
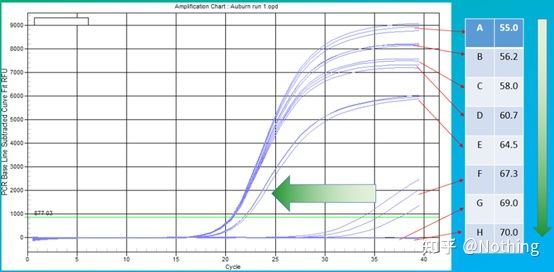
انتخاب دمای بازپخت:
· 70 درجه سانتیگراد، 69 درجه سانتیگراد - اساساً پرایمرها را نمی توان ترکیب کرد، بنابراین هیچ تقویتی وجود ندارد.
· 67.3 درجه سانتیگراد - در ابتدا مقدار کمی تقویت وجود دارد و مقدار Ct نسبتاً زیاد است.
·64.5°C——مقدار Ct کاهش می یابد.
· در 60.7 درجه سانتیگراد، 58.0 درجه سانتیگراد، 56.2 درجه سانتیگراد و 55.0 درجه سانتیگراد، مقادیر Ct اساساً به پایداری تمایل داشتند، اما مقادیر فلورسانس نهایی متفاوت بودند.
چگونه انتخاب کنیم؟اصل: اصل اول مقدار Ct بالاتر است.برای همان مقدار Ct، دمای بازپخت بالاتر را انتخاب کنید تا از دایمر شدن و تقویت غیر اختصاصی جلوگیری کنید.اگرچه مقدار فلورسانس بالاتری در دمای 55 درجه سانتیگراد وجود دارد، اما ممکن است دایمرها یا تقویت غیر اختصاصی در آن وجود داشته باشد.
اما اگر به اندازه خود باهوش باشید، قطعاً فکر خواهید کرد: از نظر منطقی، اگر واکنش PCR بسیار خاص باشد، تا زمانی که غلظت پرایمر از حداقل نیاز بیشتر باشد، نقاط بالا و پایین نباید تأثیری داشته باشند، درست مانند رنگهای فلورسنت و dNTP.در واقع، تا زمانی که دمای بازپخت به درستی بهینه شود، اثر غلظت پرایمر بر مقدار Ct به طور طبیعی به حداقل می رسد.
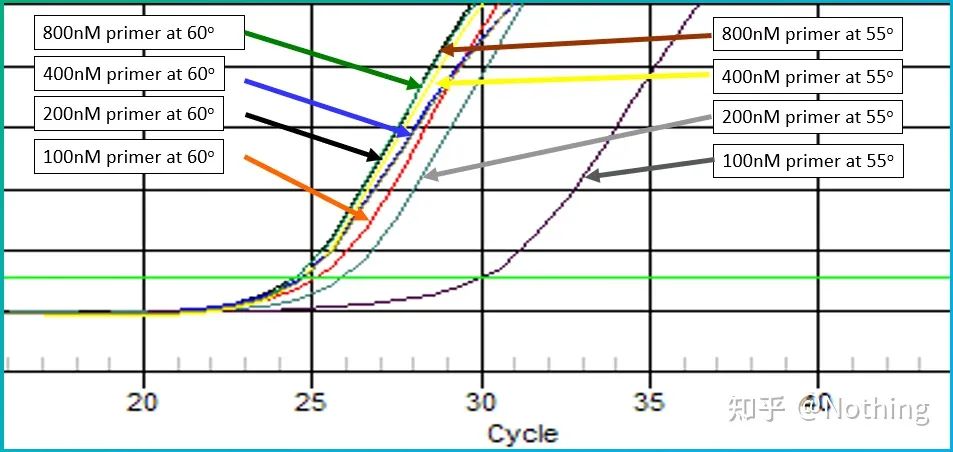
دمای بازپخت به درستی بهینه شده است و تأثیر غلظت پرایمر بر CT به حداقل می رسد
ساختار ثانویه بر راندمان تقویت تأثیر می گذارد
بیایید تصویر را از Bio-rad بگیریم تا مشکل را نشان دهیم.همچنین یک گرادیان دما برای تقویت یک ژن با ساختار ثانویه طراحی می کند.
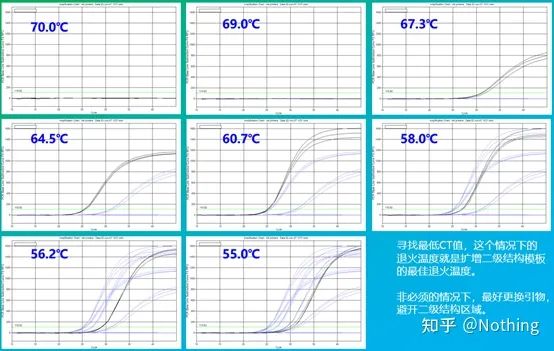
ساختار ثانویه پدیدار می شود
مشاهده می شود که با کاهش گرادیان دما، محصولات شروع به ظاهر شدن می کنند و مقدار Ct به سمت جلو حرکت می کند و به حداقل مقدار در 60.7 درجه سانتی گراد می رسد و سپس با کاهش گرادیان دما، مقدار Ct بزرگتر می شود.برعکس، با افزایش دما، ساختار ثانویه باز می شود و راندمان تقویت افزایش می یابد.پس از رسیدن به دمای معین، افزایش دما نمی تواند راندمان تقویت را بهبود بخشد.زیرا در این زمان نمی توان پرایمرها را به طور پایدار با هم ترکیب کرد.از این رو،به دنبال دما با کمترین مقدار Ct باشید، که بهترین دما برای تقویت قالب سازه ثانویه است!البته، احمق های باهوش باید بدانند که اگر لازم نیست، بهتر است پرایمرها را عوض کنند و از ناحیه ساختار ثانویه خودداری کنند.
5. سطح برنامه
MIQE - تجزیه و تحلیل داده ها

تجزیه و تحلیل داده ها عمدتاً توسط دستگاه PCR کمی فلورسنت ارائه می شود.در مقاله قبل کارهای زیادی برای تجزیه و تحلیل داده ها انجام شده است مانند کنترل خالی که در طراحی آزمایش توضیح داده شده است.ژن های مرجع داخلی، اعداد تکرار و غیره مشخص شده اند.، در اینجا به طور عمده کاربرد qPCR را توضیح می دهیم.
qPCR به طور گسترده مورد استفاده قرار می گیرد و تأیید تجربی و تشخیص اسید نوکلئیک رایج ترین سناریوهای مورد استفاده هستند.
کمی سازی مطلق
Log (غلظت اولیه) با تعداد چرخه ها رابطه خطی دارد.یک منحنی استاندارد را می توان از یک استاندارد با شماره کپی اولیه شناخته شده ترسیم کرد، یعنی می توان رابطه خطی واکنش تقویت را به دست آورد.با توجه به مقدار Ct نمونه، غلظت موجود در نمونه قابل محاسبه است.مقدار قالب هایی که باید گنجانده شود.
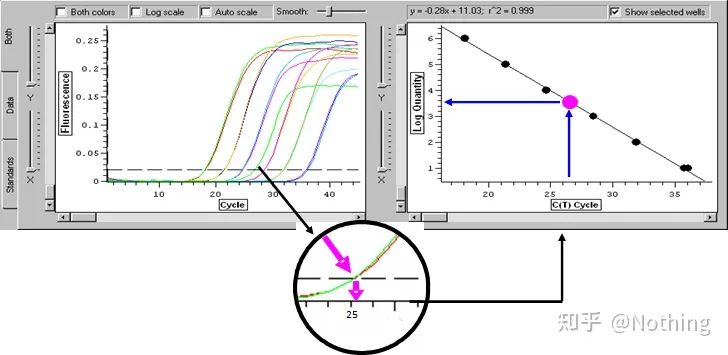
روش محاسبه کمی مطلق
کمیت مطلق باید بر اساس منحنی استاندارد باشد.برای ایجاد یک منحنی استاندارد، یک استاندارد لازم است.معمولاً استاندارد یک پلاسمید است که از شبیه سازی ژن هدف به دست می آید.چرا پلاسمید است؟زیرا DNA پلاسمید دایره ای پایدارترین است.محصول استاندارد را با توجه به نسبت دوبرابر شدن (رقت دهی) در 5 تا 6 گرادیان رقیق کنید و در هنگام رقیق شدن به یکنواختی توجه کنید.بگذارید مقدار Ct بین 15-30 باشد.
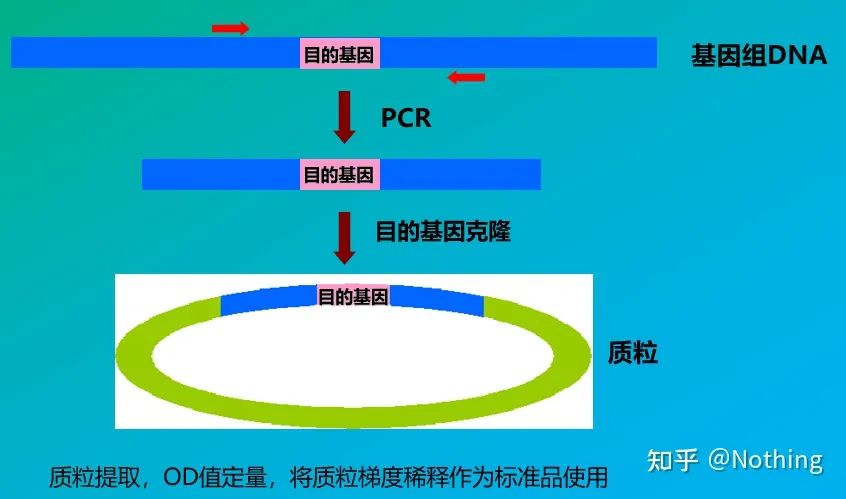
آماده سازی استاندارد
در عین حال، نمونه مورد آزمایش نیز باید بر این اساس رقیق شود (ضریب رقت را به خاطر بسپارید)، و مقدار Ct نیز باید بین 15-30 باشد.محصول استاندارد + نمونه مورد آزمایش با هم روی دستگاه قرار می گیرد.پس از اجرا، یک منحنی استاندارد با ماده استاندارد ساخته شد و نمونه های مورد آزمایش برای محاسبه غلظت به منحنی استاندارد آورده شدند.
کمیت HBV ویروس هپاتیت B یک کمیت مطلق معمولی است که می تواند تعداد کپی ویروس را در 1 میلی لیتر خون محاسبه کند.
محاسبه شماره کپی
غلظت نمونه مورد آزمایش (ng/ul) = OD260 × 50ug/ml × ضریب رقت
وزن مولکولی نمونه = تعداد بازها × 324
شماره کپی نمونه مورد آزمایش (کپی/ul) = غلظت نمونه مورد آزمایش / وزن مولکولی نمونه × 6 × 1014

روش محاسبه شماره کپی
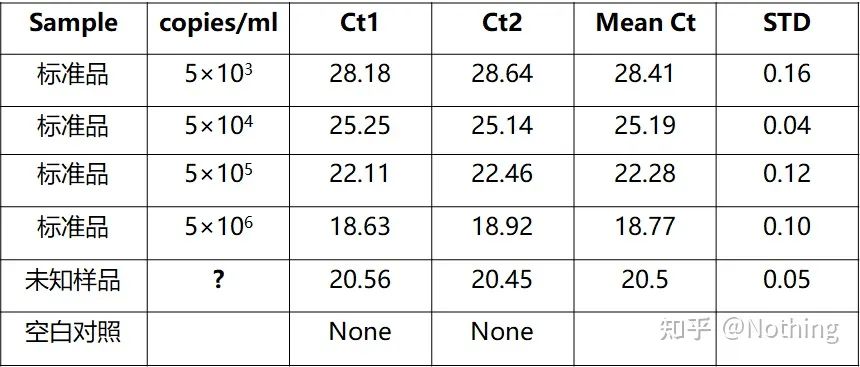

در بالا روش محاسبه برای تعیین مقدار است.این یک مسئله ریاضی است که بعد از فارغ التحصیلی از دبیرستان قابل حل است و مسائل ریاضی عموماً توسط رایانه حل می شود.اگر متوجه نشدید می توانید بیایید ارتباط برقرار کنید.
کمی سازی نسبی
کمی سازی نسبی عمدتاً در تحقیقات علمی استفاده می شود.چند ویروس در 1 میلی لیتر خون وجود دارد، و این یک ویروس DNA است، این یک رویداد نسبتاً قطعی است: مقدار خون را می توان تعیین کرد، و ویروس DNA نسبتاً پایدار است.با این حال، مقایسه تعداد نسخههای رونویسی یک ژن خاص در یک برگ برای ما دشوار است، زیرا تعیین اندازه، وزن و نرمی برگ دشوار است، تعیین مقدار RNA استخراجشده دشوار است، و تعیین کارایی رونویسی معکوس نیز دشوار است، یعنی هر مرحلهای ممکن است باعث شود که دادههای آزمایشی نتوانند اشکال داشته باشند.
بنابراین، کمی سازی نسبی باید یک عنصر را معرفی کند:ژن مرجع داخلی
به عبارت دیگر، کمیت نسبی در واقع مقایسه بین ژن هدف و ژن مرجع داخلی است.در مقایسه در همان بافت و همان سلول، تأثیر اندازه نمونه، مقدار استخراج RNA، بازده رونویسی معکوس و کارایی PCR نسبتاً کم است.به دلیل حجم نمونه کوچک، هم ژن های مرجع داخلی و هم ژن های هدف نسبتاً کاهش یافتند.به همین دلیل است که ما قبلا بر یکنواختی و ثبات تاکید کرده ایم.
ژن های مرجع داخلی به طور کلی هستندژن های خانه داری(ژن های خانه دار)، که به دسته ای از ژن ها اطلاق می شود که به طور پایدار در همه سلول ها بیان می شوند و محصولات آنها برای حفظ فعالیت های اساسی حیات سلول ها ضروری است.
این مفهوم را اشتباه نگیرید.ژن های خانه داری اصطلاحات عملکرد بیولوژیکی هستند، در حالی که ژن های مرجع داخلی اصطلاحات فنی تجربی هستند.ژنهای خانهدار قبل از انتخاب بهعنوان ژن مرجع داخلی نیاز به تأیید اعتبار دارند.
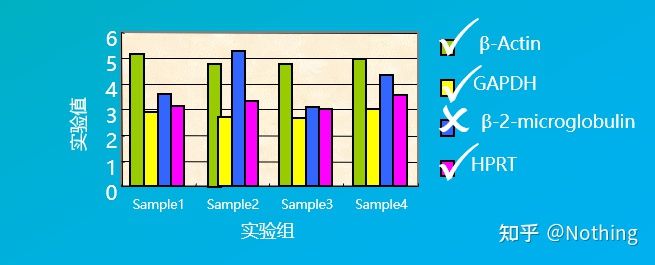
به عنوان مثال، ما چندین ژن خانه دار را در شکل زیر برای آزمایش سطح بیان آنها در سلول های بافتی مختلف انتخاب کردیم و دریافتیم که سطح بیان β-2-میکروگلوبولین کاملاً متفاوت از سه ژن دیگر است، بنابراین نمی توان از آنها به عنوان ژن مرجع داخلی استفاده کرد.
پس از درک عملکرد تصحیح ژن مرجع داخلی، با توجه به معرفی ژن مرجع داخلی، دو الگوریتم استخراج می شود.
· روش منحنی استاندارد دوگانه
·2 – △△روش Ct (روش مقایسه مقدار CT)
اگر به مطالعه گونه ها و توابع ژنی علاقه مند هستید، لطفاً تحقیق در مورد الگوریتم ها را رها کنید و مستقیماً از فرمول ها استفاده کنید یا مستقیماً از ماشین ها استفاده کنید.اگر در ریاضیات و مهندسی یک مرد مستقیم هستید، لطفا احساس راحتی کنید.
روش منحنی استاندارد دوگانه
ژن هدف و ژن خانه داری نمونه شاهد و نمونه مورد آزمایش را از طریق منحنی استاندارد کمی کنید و سپس مقدار نسبی را طبق فرمول محاسبه که سطح بیان نسبی است محاسبه کنید.
مزایا: تجزیه و تحلیل ساده، بهینه سازی تجربی نسبتا ساده
عیب: برای هر ژن، هر دور آزمایش باید یک منحنی استاندارد ایجاد کند
کاربرد: یکی از دو روش کمی نسبی رایج و شناخته شده در مطالعه تنظیم بیان ژن
فرمول به شرح زیر است:

نمونه ها به شرح زیر است:
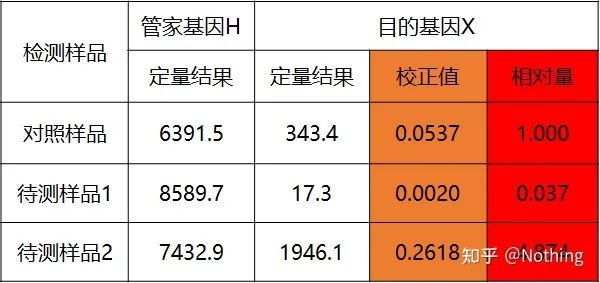
مقدار نسبی را بر اساس نتیجه کمی محاسبه کنید
2 – روش △△Ct (روش مقایسه مقادیر CT)

مزایا: نیازی به ایجاد منحنی استاندارد نیست
معایب: فرض بر این است که راندمان تقویت نزدیک به 100٪ است.انحراف استاندارد کمتر از 5% است و منحنی استاندارد و راندمان بین هر تقویت ثابت فرض می شود.بهینه سازی شرایط تجربی پیچیده تر است.
کاربرد: یکی از دو روش کمی نسبی رایج و شناخته شده در مطالعه تنظیم بیان ژن
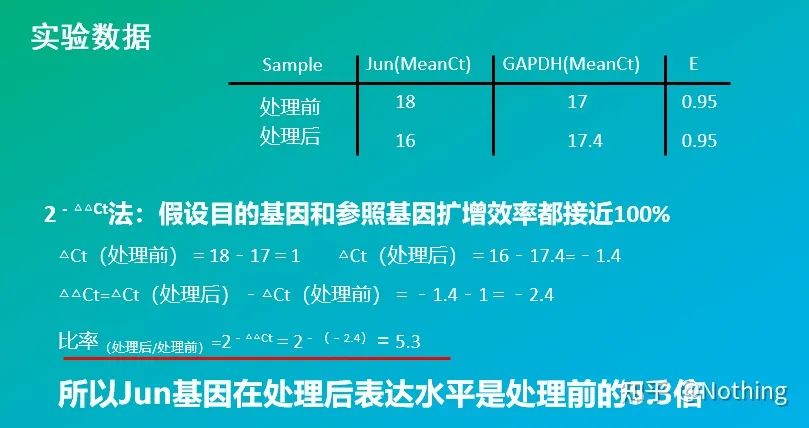
البته راندمان تکثیر معمولاً غیرممکن است که به طور کامل 1 باشد. روش اصلاح: اگر بدانیم که ژن هدف و ژن مرجع دارای راندمان تقویت یکسانی هستند اما بازده تقویت برابر با 1 نیست، می توان 2-△△Ct را به صورت زیر تصحیح کرد: ، سپس فرمول محاسبه را می توان به 1.95 - △△Ct اصلاح کرد
تا کنون، مطالب مربوط به PCR کمی فلورسنت به پایان رسیده است.
زمان ارسال: آوریل-06-2023



